 電子發燒友App
電子發燒友App


傳統鋰離子電池(LIB)在能量密度方面遇到了應用瓶頸,而新興的鋰硫(Li-S)電池在下一代可充電電池系統中顯示出可實現更高能量密度的潛力,這是由于硫正極(1675 mAh g?1)和金屬鋰負極(3860 mAh g?1)均具備相當高的理論比容量。此外,結合輕量化、環境友好和低經濟成本等優點,鋰硫電池在過去十幾年中已成為最有前途的電池體系之一,并得到不斷發展。然而,這些研究工作主要是在室溫(RT)下進行的。由于鋰硫電池的電化學反應過程相對緩慢,在低溫(LT)情況下所進行的相關研究仍較少。但對于實際情況來說,如電動車輛、空間/極地探索、水下作戰和邊境國防等場景,迫切需要能適應寒冷環境的高比能可充電電池體系。
【工作介紹】
吳飛翔教授團隊前期的工作(Nano Lett. 2020, 20, 5391.)已經證明采用低濃度電解液可提高室溫鋰硫電池的循環容量保持率和實現高達2 C的優異倍率性能。這主要是由于低濃度電解液具有低粘度、更好的潤濕性以及在常溫下對多硫化物穿梭的抑制。基于此,中南大學吳飛翔課題組等人于近日采用低濃度電解液(LCEs)來提升鋰硫電池在惡劣條件下的電化學性能,包括低溫工況、普通硫材料和高載量正極。發現即使在低溫條件下LCEs也能提供更高的容量和更高的容量保持率。更重要的是,利用低濃度電解液策略對鋰硫電池在低溫下的電化學和動力學行為進行了深入研究。這些發現和見解將揭示低濃度電解液在實現低溫鋰硫電池具備更快動力學的作用。此外,低濃度電解液這種方法可為實現更實用的低溫鋰硫電池提供了一種實際可行的方法。該文章發表在國際材料領域頂級期刊《先進功能材料》(Adv. Funct. Mater.)上。通訊作者為中南大學吳飛翔教授和湖南大學柳斌副教授;中南大學19級博士生褚福路為本文第一作者,湖南大學19級碩士生王萌為共同第一作者。
【內容表述】
鋰硫電池主要是基于非拓撲轉換化學,放電過程基本上有兩個步驟(即兩個不同的電壓平臺)。位于高電壓區(從2.5到2.3 V)的第一個平臺對應于硫轉化反應,該反應過程會經歷硫環的斷裂,形成可溶于醚類電解液的長鏈硫組分即多硫化物(LiPSs),隨后會產生難以控制的穿梭效應。另一個平臺位于較低電壓范圍(≈2.1 V),符合從短鏈多硫化物到Li2S2/Li2S的轉化。實際上,每個充放電循環都是一個復雜的多相電化學過程。因此,在硫氧化還原反應過程中,可實現完全轉化反應的快速動力學是至關重要的。然而,現實情況中從Li2S4到Li2S的完全轉化很難實現,轉化反應動力學非常緩慢。再加上最終放電產物Li2S的絕緣性能,這種情況在低溫工況下會加劇惡化。因此,為了成功實現鋰硫電池的低溫性能,加速轉化反應動力學特別是在Li2S4到Li2S轉化過程中的轉化動力學變得非常重要。
本文采用低濃度電解液成功實現了在低溫環境中(在0和?20 °C)加速鋰-硫轉化反應速度,尤其是從Li2S4到Li2S過程中的轉化動力學,并抑制了多硫化物的穿梭效應,從而有效提升了低溫鋰硫電池的性能。這主要是由于低濃度電解液的低粘度、更好的潤濕性,以及穩定的電極界面層進而對多硫化物穿梭有效抑制。原位EIS和CV等電化學測試手段揭示了0.1 M和1 M電解液中電極動力學的主要差異,進一步解釋了兩種電解液在工作機理上的差異。對循環后的電極進行的界面化學分析表明,在LCEs體系中構建了以有機物為主和一些有利無機物組分共存的混合界面層,進而展現出較小的表面層電阻。這些發現闡明了LCEs在實現低溫鋰硫電池提升反應動力學方面的作用,并為在極端條件下實現高性能鋰硫電池提供了簡單、低成本和廣泛適用的途徑。
1. 低溫鋰硫電池在低濃度電解液中的電化學性能
為了顯示出低濃度電解液的最大改善效果,直接使用商業大顆粒硫粉與常規導電劑石墨烯混合得到石墨烯-硫(G–S)復合物,而無需任何功能性基體或特定催化位點。如圖1所示,在0°C環境下評估使用G-S正極材料(面積硫負載量:1.0 mg cm-2)的鋰硫電池的電化學性能。可以看到,使用0.1 M電解液時,電池在不同電流密度下均呈現出比常規電解液(1 M)高得多的可逆容量(圖1a-c),并具有更正常的放電/充電曲線(圖1d-i)。
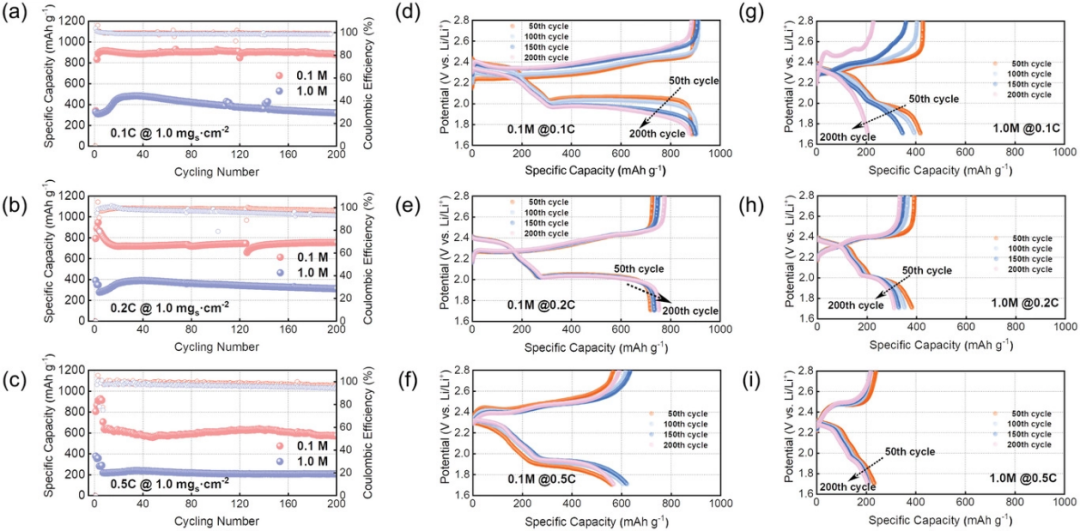
圖1. 石墨烯-硫(G–S)正極在0℃條件下在0.1和1.0 M電解液中不同倍率下的電化學循環性能:a)0.1 C、b)0.2 C、C)0.5 C,以及d–i)隨循環圈數變化的相應容量-電壓曲線圖。
同樣的,在不同循環次數和不同倍率條件下的充放電曲線(圖1d–i和圖2a)進一步加深了對電解液濃度及其可逆容量之間聯系的理解。使用LCEs的電池保持著良好的充放電曲線形狀,在不同的倍率下具有明顯更高的放電容量和更小的電壓滯后,可為在低電壓范圍內的短鏈多硫化物到不溶性硫化物的轉化反應提供了更好的動力學。而對于使用1 M電解液的電池,其總容量主要來源于第一個放電平臺,而對應于短鏈多硫化物到固態硫化物轉化的第二個放電平臺似乎愈發不清晰可見(圖1g-i)。即使在G–S硫正極的面載量進一步增加時,具有LCEs的電池也顯示出良好的循環性能(圖2a-c)。
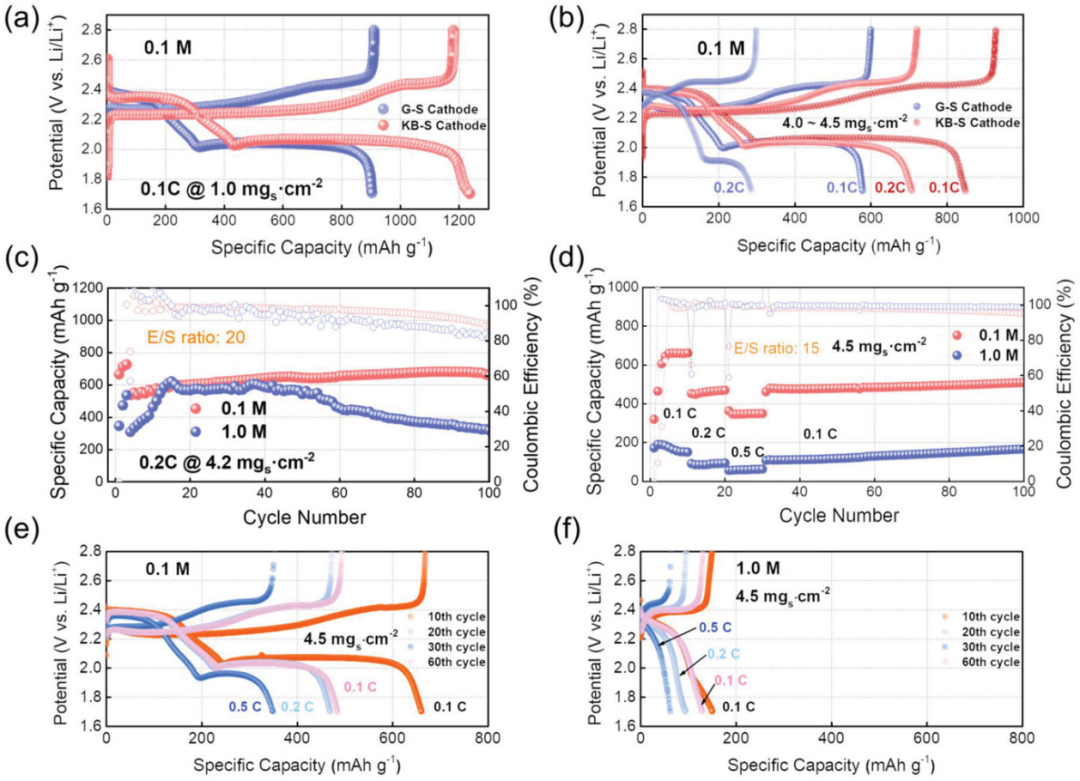
圖3. 具有不同載量的石墨烯-硫(G–S)和科琴黑-硫(KB-S)正極在0°C下的電化學性能比較研究:a)1.0 mg cm-2和b)4.5 mg cm-2?使用0.1和1.0 M電解液的高載量KB-S正極的c)循環穩定性和d)倍率性能。使用e)0.1 M和f)1.0 M電解液進行倍率測試所對應的充放電曲線圖。
由于商用硫塊直接用于簡單的G-S正極,幾十微米的顆粒可能會限制硫的容量利用率。因此,為了進一步驗證低濃度電解液的有效性和普遍性,又選擇了通過常用的熔融-浸漬法得到的納米級科琴黑-硫(KB-S)復合正極,來研究低溫下的鋰硫電池。為了詳細探討LCEs在更實際條件下的有效性,進一步研究了高載量KB–S樣品(圖3b-f)。特定循環下充電和放電平臺之間的電壓差表明,在0.1 M電解液中循環時,電壓滯后增長較小。相比之下,使用標準濃度電解質(SCE)的電池最初顯示出相當低的容量和較長的活化時間,且其容量很快下降到低得多的容量以及庫倫效率(CE)的波動遠大于LCE(圖3c)。這應該是由于溶解的多硫化物的更嚴重穿梭行為和低導電含硫物質的進一步沉積而導致的活性電極失活,使用高負載正極會加劇這種情況。當進一步降低電解液的用量(E/S=15 μL mg–1)時,0.1 M體系在不同的倍率下提供了與1.0 M相比具有競爭力的容量(圖3d)和良好的重復性。圖3e,f中的相應電壓曲線顯示,1.0 M電解液中鋰-硫轉化反應行為不完全,表明該電池中的反應動力學相當差。
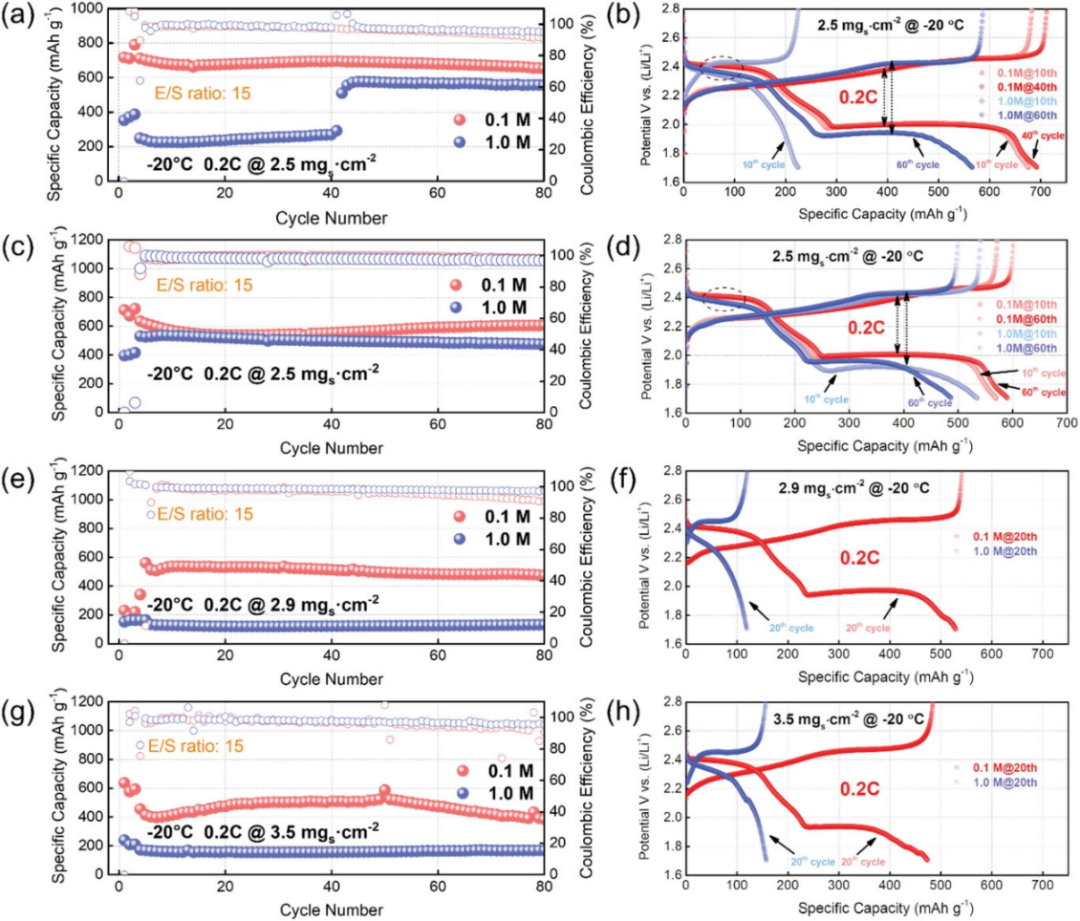
圖4. 在-20℃條件下,不同載量KB–S正極在0.2 C下使用0.1和1.0 M電解液的循環性能及其相應的電壓曲線圖:在2.5mg cm–2載量條件下,a,c)循環性能和b,d)相應的電壓曲線圖的平行測試。
在面向實際應用的更苛刻的條件下即?20℃,對低溫鋰硫電池使用0.1 M電解液的可行性進行了詳細評估。圖4顯示了使用兩種電解液的電池在?20℃下的循環性能和相應的電壓曲線。在這樣的低溫下,電池中的硫氧化還原反應速率不可避免地變得非常慢。S8轉化為長鏈多硫化物的轉化反應或許可能實現,但從Li2S4直接轉化為固體Li2S2或甚至Li2S將變得相當困難。如圖4a所示,使用SCE的KB-S正極無法在0.2 C下完成固相轉化,提供了非常低的初始容量。再加上1 M電解液的潤濕性較差,典型的電壓曲線顯示幾乎消失的第二電壓平臺,進一步證實了在該低溫下硫氧化還原反應動力學的嚴重惡化(圖4b,第10個循環)。而對于在0.2 C下使用LCE的電池,它釋放出711 mAh g-1的高放電容量,提供清晰的具有較小極化的第二電壓平臺(圖4a,b)。當繼續將硫載量增加,LCE仍然可以保持其競爭優勢(圖4e,f)。采用稀溶液的電池在0和?20°C下,均可實現短鏈多硫化物向固體硫化物的過渡,并具有更好的一致性,而常規電解質中的電化學性能均勻性較差。這可以得出結論,在低溫下使用SCE的內部動力學是不穩定和變化的,并且通常需要長時間的活化,以維持較高載量和貧電解質條件下的硫利用率,甚至活化失敗(圖4e-h)。
為了研究使用不同電解液電池的詳細電化學行為,在低溫環境(0和?20℃)中進行了循環伏安法(CV)測量以探測LCEs的積極影響。伏安曲線分別如圖5所示,顯示了低濃度溶液對硫化學反應動力學的明顯影響。具有低濃度電解液的電池在0℃下顯示出兩個顯著的峰值(圖5a),這可歸因于固體硫還原為長鏈多硫化物,并進一步得到最終放電產物,如Li2S2/Li2S。對于反向掃描中的氧化峰,它們歸因于最終放電產物(Li2S)向中間產物(LiPSs)的可逆轉化,然后轉化為活性硫。相比之下,具有常規電解液的電池具有較大的極化和較小的峰面積。從第二個循環開始,氧化還原峰的位置和面積略有變化,表明反應可逆性和循環穩定性良好(與常規體系相比,峰面積更大,曲線重疊,極化更小)。注意,首圈還原反應的明顯位置變化應歸因于初始循環中的電池活化。
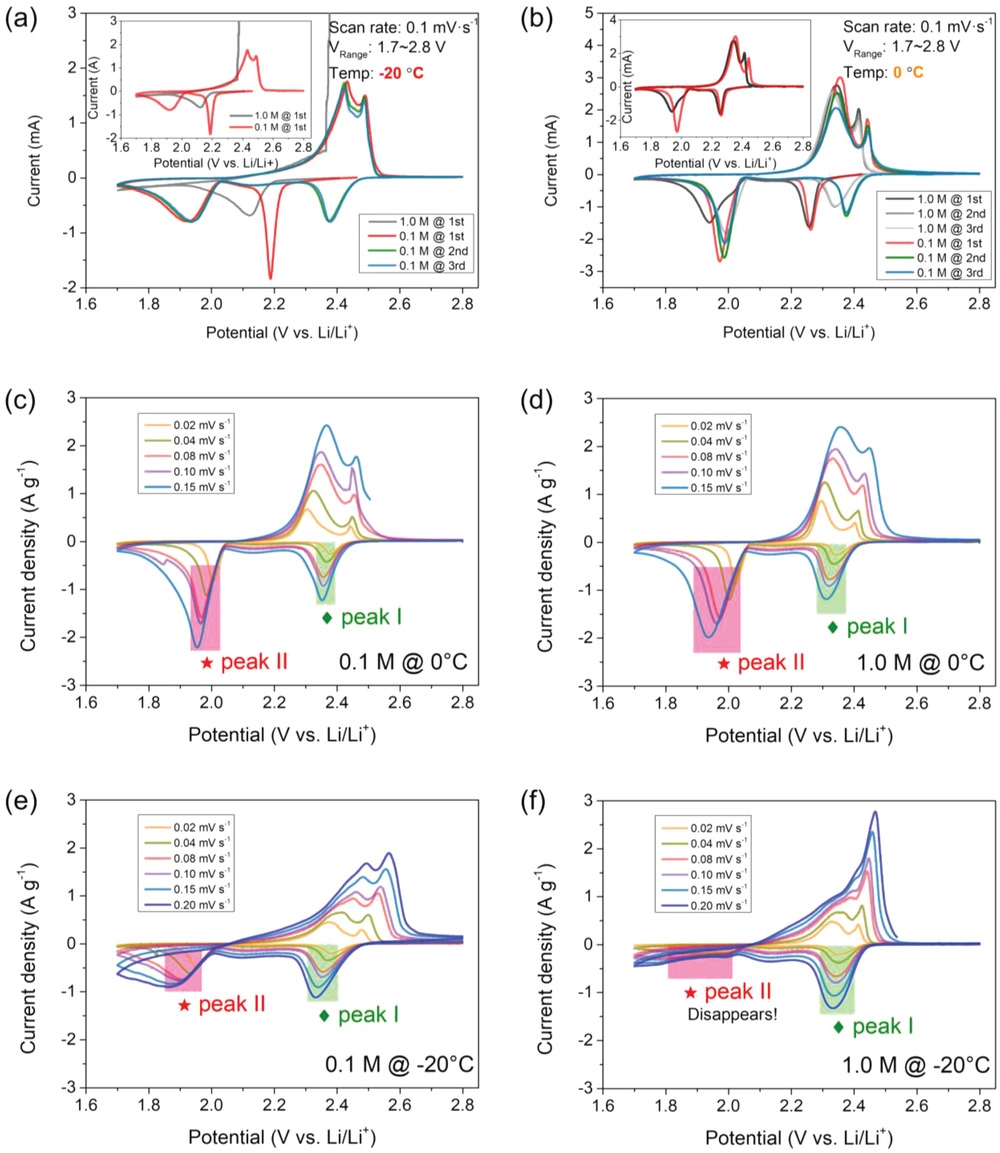
圖5. 使用不同電解液的低溫Li–S電池的循環伏安曲線:在0.1 mV S-1下使用0.1和1.0 M電解液在a)0和b)?20 ℃環境進行重復循環伏安掃描;使用不同電解液的低溫鋰硫電池在不同掃描速率下的循環伏安曲線(ν=0.02、0.04、0.08、0.10和0.20 mV S?1),c)0℃下0.1 M,d)0 ℃下1.0 M,e)-20 ℃下0.1 M和f)-20 ℃下1.0 M。
在?20℃時,這些電池的電化學行為差異更為明顯。如圖5b所示,1 M電池經歷了有限的硫氧化還原反應,在初始掃描期間,僅在2.12V下顯示了單個還原峰,峰面積較小,第二還原行為(低于2.0V)幾乎消失。因此,由于不存在多硫化物被還原為最終產物Li2S的行為,鋰化動力學受到極大限制。此外,1 M電池在正掃期間經歷了大量極化的過充電行為(圖5b插圖),這是由于LiPSs的不可控穿梭損耗造成的。但LCE電池在圖5b中表現出完全不同的電化學行為。在第一個循環中,可以在2.19和1.928V處發現明顯的還原峰。此外,在2.433和2.49V下,兩個氧化峰也清晰可見。在隨后的循環中,即使在?20℃電池的陽極和陰極峰很好地重疊,表明在硫轉化反應期間優越的循環穩定性和更好的可逆性。
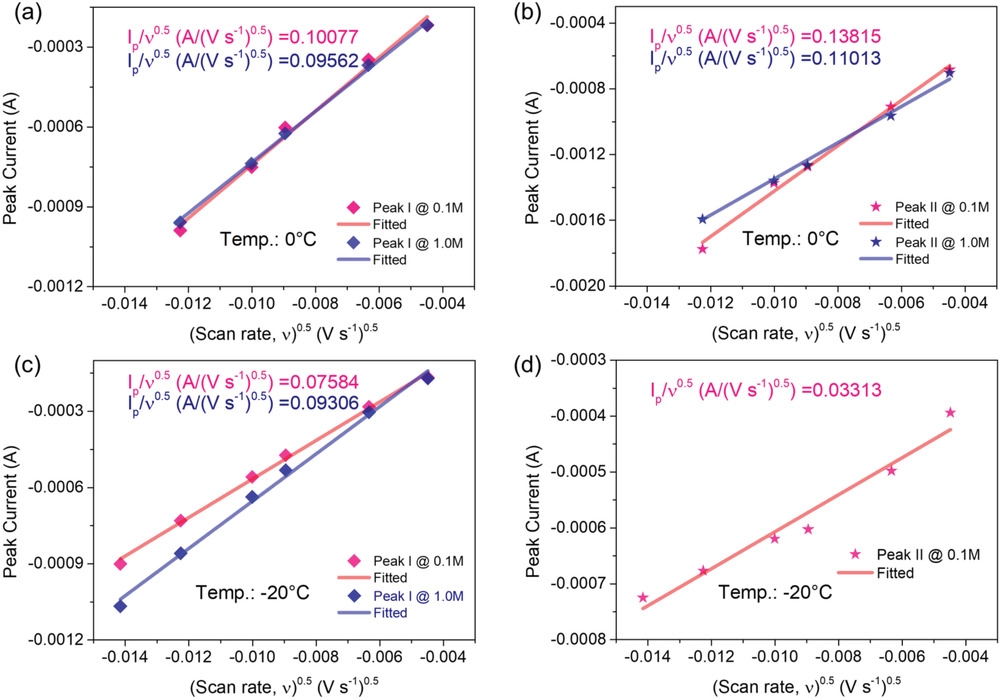
圖6. CV曲線的各種峰值電流與掃描速率平方根的關系圖,以及從Randles–Sevcik分析獲得的線性關系擬合。a)0 ℃下的峰值I,b)0 ℃下的峰值II,C)?20 ℃下的峰值I和d)?20 ℃下的峰值II。
此外,在硫氧化還原反應期間,鋰離子Li+在電解液和電極之間界面上的擴散動力學通過一系列CV曲線進行評估(圖5和6)。圖5c-f中的這些結果表明,在相對較低的掃描速度下,硫轉化具有良好的氧化還原反應行為。另一個令人印象深刻的方面是,在?20℃(圖5f)使用1 M電解液的電池其第二還原峰消失,強烈表明當使用LCE時,多硫化物的轉化動力學在低溫下得到加速。如圖6所示,峰值電流(ip)與掃描速率的平方根(η1/2)呈良好的線性關系,LCE比常規LCE更陡,這意味著使用稀溶液的電池具有更好的動力學。然后,根據Randles–Sevcik方程計算放電過程各階段的鋰離子擴散系數(DLi+)。對于具有LCE的系統,在低溫條件下,DLi+值顯著優于對照組(1.0 M電解液),例如LCE的擴散系數(峰值I在?20℃)為≈7.1×10–7 cm2?s?1明顯高于常規體系(≈1.4×10–9 cm2?s?1). 總之,這些結果令人信服地證明了低濃度電解液在促進反應動力學和規范電極表面界面行為方面的重要性。
2. 低濃度電解液的物化屬性
低濃度(0.1 M)體系提供了非常低的粘度以釋放高的鋰離子遷移率。0.1 M電解液在25, 0, ?20 ℃分別顯示出1.83、1.816和1.63 mS cm-1(圖7a),而1.0 M電解液中對應數值為12.14、11.09和8.52 mS cm?1。盡管環境溫度不同,LCE的離子電導率幾乎不變,可以滿足鋰硫電池的正常工作。值得注意的是,在一定電流密度范圍內,活性離子(Li+)的相間溶劑化/脫溶劑化和隨后的遷移遠比離子電導率的值重要。因此,稀釋液中足夠的離子電導率可以滿足各種硫正極在寒冷環境中的高性能,如先前數據所示。此外,使用接觸角計評估它們對硫正極的潤濕性。各種硫正極的靜態接觸角如圖7b所示。對于G–S正極,LCE中的接觸角(CA)僅為13.5°,而SCE樣本顯示為21.3°。對于KB–S正極,與使用1.0 M電解液時的接觸角(25.4°)相比,LCE的接觸角也較小,為11.9°,表明低濃度溶液的高潤濕性。因此,正極上良好的潤濕性和低濃度的適當離子導電性可以加速電極反應過程,從而即使在高倍率下也能提供更高的容量保持率和更低的過電位(圖1-4)。
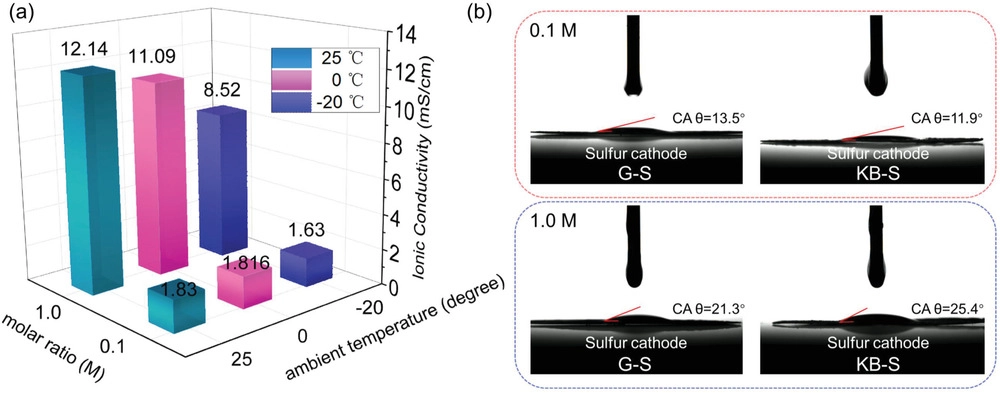
圖7. 0.1和1 M電解液的物理性質:a)不同溫度下的離子電導率;b) 通過接觸角測量在硫正極上的相應潤濕性。
3. 低溫鋰硫電池中循環后電極的形貌演化

圖8. 在0℃下運行的鋰硫電池中循環電極的形態特征:在a、b)0.1 M和C、d)1.0 M電解液中循環500次后,G-S正極的表面SEM圖像;在e,f)0.1 M和g,h)1.0 M電解液中循環500次后鋰金屬負極的SEM圖像。(d)中的黃色虛線圓圈表示多硫化物的不均勻穿梭沉積。
使用掃描電子顯微鏡(SEM)來觀測循環后電極上的表面形貌變化。如圖8a,b所示,在0.1 M電解液中循環的正極其納米結構完整性可以很好地保持,同時有最小的裂紋和較少顆粒粉碎。平坦表面形貌表明多硫化物沉積更均勻,多硫化物在0.1 M電解液中的擴散可控。由于短鏈多硫化物(Li2S2)傾向于在0.1 M電解液中聚集(較難溶解),長鏈多硫化物和Li2S2之間的化學結合可以成功地抑制多硫化物的穿梭效應。相反,常規體系循環正極的表面顯示出更多的孔、裂紋和鋰多硫化物的不均勻穿梭沉積(圖8d中的黃點圓圈),表明在低溫下,1 M電解液中存在嚴重的多硫化物穿梭效應、不良的反應動力學和有限的固-固轉化。可溶性多硫化物很難轉化為固體硫化物,導致更粘稠的電解質和這種可溶性多硫化物不受控制的擴散。因此,循環正極的形貌變得越來越粗糙,有更多的孔和裂紋(圖8c,d)。在1.0 M電解液中循環的鋰負極顯示出相同的特征,證明了可溶性多硫化物對原始鋰金屬的嚴重侵蝕。隨著鋰負極上的鈍化表面和任意位置的體積變化,循環鋰片出現更粗糙的區域,具有垂直的鋰枝晶,并形成金屬鋰基體的表面粉化(圖8g,h),表明循環期間鋰沉積不均勻。有趣的是,0.1 M電解液中的循環鋰負極(圖8e,f)顯示出均勻且高度均勻的表面,其中包含有序排列且表面形貌光滑的大鋰金屬顆粒。更重要的是,沒有任何可見的硫化物沉淀和有害的鋰細絲(即鋰枝晶)。這意味著在低溫下,0.1 M電解液成功地抑制了多硫化物穿梭效應和電解液/鋰負極界面問題。
4. 低溫鋰硫電池中低濃度電解質實現快速動力學的機理
復雜的硫氧化還原反應動力學可以從表觀電化學阻抗中得到根本反映。具有SCE和LCE的電池均在低溫環境(0℃)中進行循環,然后在不同的放電或充電深度下進行原位電化學阻抗譜(EIS)測試(見圖9)。通常,EIS曲線主要包括從高頻擾動信號到低頻的三個部分。即內阻(Rs)、界面阻抗(Ri,存在于高頻至中頻區域的半圓)和擴散阻抗(Warburg阻抗)。其中,界面阻抗是本研究的主要焦點,涉及固體電解質界面(SEI)層(RSEI)的電阻和電荷轉移電阻(Rct)。如圖9所示,在鋰硫電池的初始狀態下,界面阻抗通常顯示為單個融合半圓,表明常規系統中Rs和Ri的電阻較小,這是由于1.0 M電解液的較高離子電導率所致。隨著放電過程從開路電位(OCP)變為2.05 V,硫正極經歷固-液相轉化的第一個電壓平臺,以釋放具有更高粘度的可溶性長鏈多硫化物中間體(Li2Sn,4≤n≤8)。這些中間體將遇到電極來形成界面層,從而引起穿過電極/電解液界面的相應電荷轉移過程。因此,在高頻到中頻范圍內出現了兩個半圓。由于長鏈多硫化物的形成是通過固液相反應進行的,因此界面傳質過程將得到改善。這是在2.25 V的第一次放電中兩種電解液體系的阻抗明顯降低的主要原因。此外,可溶性長鏈多硫化物的形成將不可避免地增加電解液的體相粘度。正如先前的研究表明,LCE(0.1 M)提供了極小的粘度,這有助于高鋰離子遷移率。因此,0.1 M電解液中溶液粘度的增長也相對較小。對于LCE,作為預期結果,其在第一次放電平臺期間的電荷轉移阻抗則要更小。
在放電范圍≈2.05–1.7 V內,更多的多硫化物將參與表面構筑。加上電極上更好的潤濕能力,低濃度體系中的SEI層電阻(RSEI)將逐漸降低并穩定。在該范圍內,放電對應于短鏈多硫化物的還原過程,即從液體LiPSs中間體形成固體Li2S2/ Li2S。在2.05–1.7 V范圍內的主要差異是,稀釋溶液中的電荷轉移阻抗(Rct)遠大于標準溶液中的阻抗。這種有趣的現象也出現在充電電壓范圍≈1.7–2.3 V。根據我們之前的發現,添加LiTFSI鹽可以增強Li2S2的溶解,這意味著1 M具有更強的溶解Li2S2能力。然后,1 M電解液中的可溶性Li2S2降低了電池在固-固范圍內的Rct。更重要的是,在該固-固反應范圍內(≈?2.05–1.7和1.7–2.3 V),Li2S2在0.1 M電解液中的聚集將與電解質中的長鏈多硫化物化學鍵合,以減少穿梭效應,部分導致0.1 M電解液中更好的循環穩定性(圖1-3)。
在≈2.3–2.8 V的充電范圍內時,由于長鏈多硫化物的形成,兩種電解液中電池的Rct都大大降低。就圖9所示的第二個循環而言,總體而言,兩個電池顯示出與第一個循環相同的界面阻抗變化行為。它們的總值略有降低,這可能是由表面層的電化學活化和穩定重建所導致的。一個區別是使用0.1 M電解液的電池其整體界面阻抗降低得更明顯。另一個區別是,除2.05–1.7和1.7–2.3 V范圍外,在0.1 M電解液中大多數電壓范圍內的電荷轉移電阻(Rct)要小。
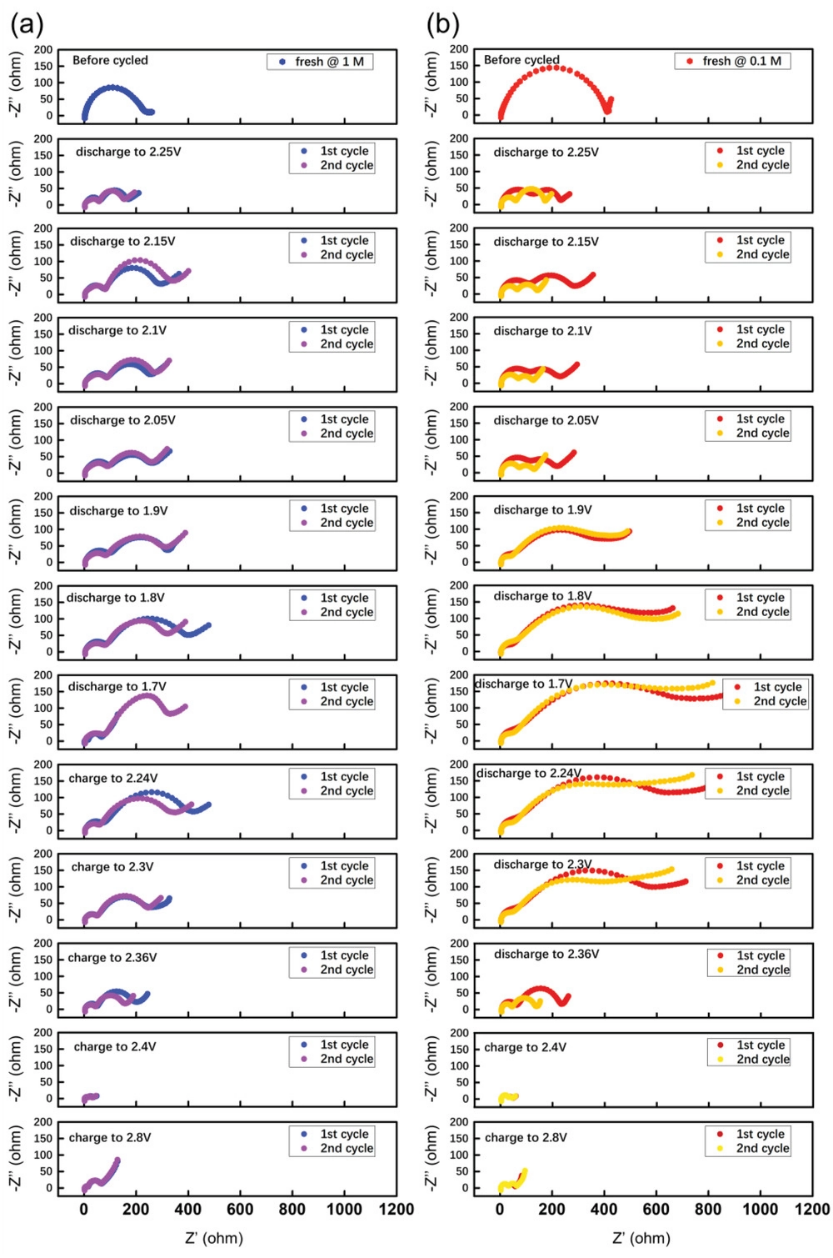
圖9. 在a)1.0 M和b)0.1 M電解液中,在0℃下,鋰硫電池在不同放電和充電狀態下的原位EIS測量。
于1 M電解液體系,這是由于低濃度電解液的以下影響:i)更好的潤濕性能,ii)可溶性多硫化物的粘度較低,iii)可能存在弱溶劑化效應,導致反應能壘較低,以適應快速電荷轉移過程。由于鹽濃度的巨大差異,預計兩種電解質體系的溶劑化結構不同。與1 M液體相比,低濃度溶液顯示出更少的Li+-陰離子相互作用組分,但更多的Li+-溶劑配位絡合物。由于不同離子絡合物的結合能明顯不同,這些溶劑化合物的去溶劑化難度不同。因此,在低濃度電解質中具有較弱結合能的高比例Li+-溶劑組分導致Li+離子去溶劑化更容易并進一步遷移穿過電極。因此,在低溫鋰硫電池中,由此產生的更快的動力學可以加速更快的電荷轉移過程,并實現更出色的倍率性能。這些結果與電化學性能一致,表明低濃度電解液能夠在極低溫度條件下實現不受阻礙的轉化反應動力學,并抑制鋰硫電池中多硫化物的穿梭,從而在各種倍率下實現相對較高的容量利用率和更好的循環穩定性。
5. 電解液/電極界面處的界面化學行為
由于鋰硫電池的快速動力學和優異的倍率能力也極其依賴于穩定的界面層,因此采用X射線光電子能譜(XPS)技術進行了表面成分分析和探究相關的化學鍵合情況。詳細總結了LCE(圖10a–c,g–i)和SCE(圖10 d–f,j–l)中形成的電極界面(SEI和正極電解質界面,CEI)的光譜情況,可發現鋰金屬負極和硫正極兩個樣品的XPS光譜(特別是C 1s、F 1s和S 2p光譜)存在明顯的差異。
在負極側,0.1 M電解液體系的C 1s光譜結果(圖10a)顯示了一個明顯的峰應歸因于C-C組分以及幾乎不可見的C-F峰。對于其他C 1s峰,可以分別對應于Li2CO3和C-O。但明顯的C-OR(R為烷基)峰在常規體系中顯現出來,而其他碳峰是正常強度(圖10d)。這些碳基組分的強度越強,表明各個SEI層的組成差異越大,特別是在LCE體系中以C-C和-CO3有機組分為主的外部SEI層。這一趨勢可以廣泛驗證,即SEI層由無機和有機組分組成,外層提供更多的有機組分,如-COOR,-CxHyOz,and-CO3。更特別的是,對于低濃度的情況,鹽與溶劑的低摩爾比易于誘導有機占主導地位的界面層,因為大量溶劑會優先得到還原。C-F峰證明了稀釋電解液中副反應(消耗鋰鹽)的減少,因為相關官能團-CF3最有可能來自LiTFSI鹽中陰離子的分解。這一現象也可以在F 1s光譜中顯示,其中一個極弱的峰即C-F出現在0.1 M電解液中(圖10b)。
僅就峰值強度而言,低濃度體系中F和S元素的光譜強度略低,這也是因為它們的鹽與溶劑的摩爾比低。但是在每一表面層中所得物種之間的比例相當不同。由LCE得到的表面層中LiF的高含量應源自LiTFSI分解。根據之前的研究,在低濃度的情況下,LiTFSI大部分被解離,這可能有助于產生更多的還原產物。LiF作為SEI層中眾所周知且幾乎無處不在的成分,由于高楊氏模量和低擴散能壘,可以提供更好的SEI層,以實現穩定性和快速離子擴散。因此,使用LCE可以獲得具有良好平整度的堅固SEI層(通過SEM證明),從而提高倍率能力。而對于其余組分,在低濃度存在下,誘導LiCF3和Li2NSO2CF3的副反應被抑制。因此,在LCE體系的初始循環期間,LiTFSI主要分解為LiF,并進一步伴隨著不良反應的減少(低占比的C-F組分)作為已經穩定的界面。此外,由于硫氧化還原的快速動力學和完全放電過程,在0.1 M電解液中可以看到更多的硫基物質(圖10c)。Li2Sx的存在表明Li金屬與可溶性多硫化物之間發生化學反應。LCE中多硫化物的受控穿梭效應將導致鋰金屬負極的有限腐蝕(更少的Li2Sx組分)。從包括Li、N和O元素的其他光譜(圖S9,支持信息),可以發現類似情況。大量研究表明,氮化界面可以促進鋰離子的快速擴散,LiNxOy和鋰多硫化物的協同效應可以調節表面形貌和均勻的鋰離子通量分布。因此,低濃度電解質可以形成具有更多有機成分且不缺乏堅固無機物的混合SEI層。這種穩定的界面層可以將鋰金屬負極與電解液隔離,并確保快速離子擴散(更好的倍率性能)和更均勻的表面。
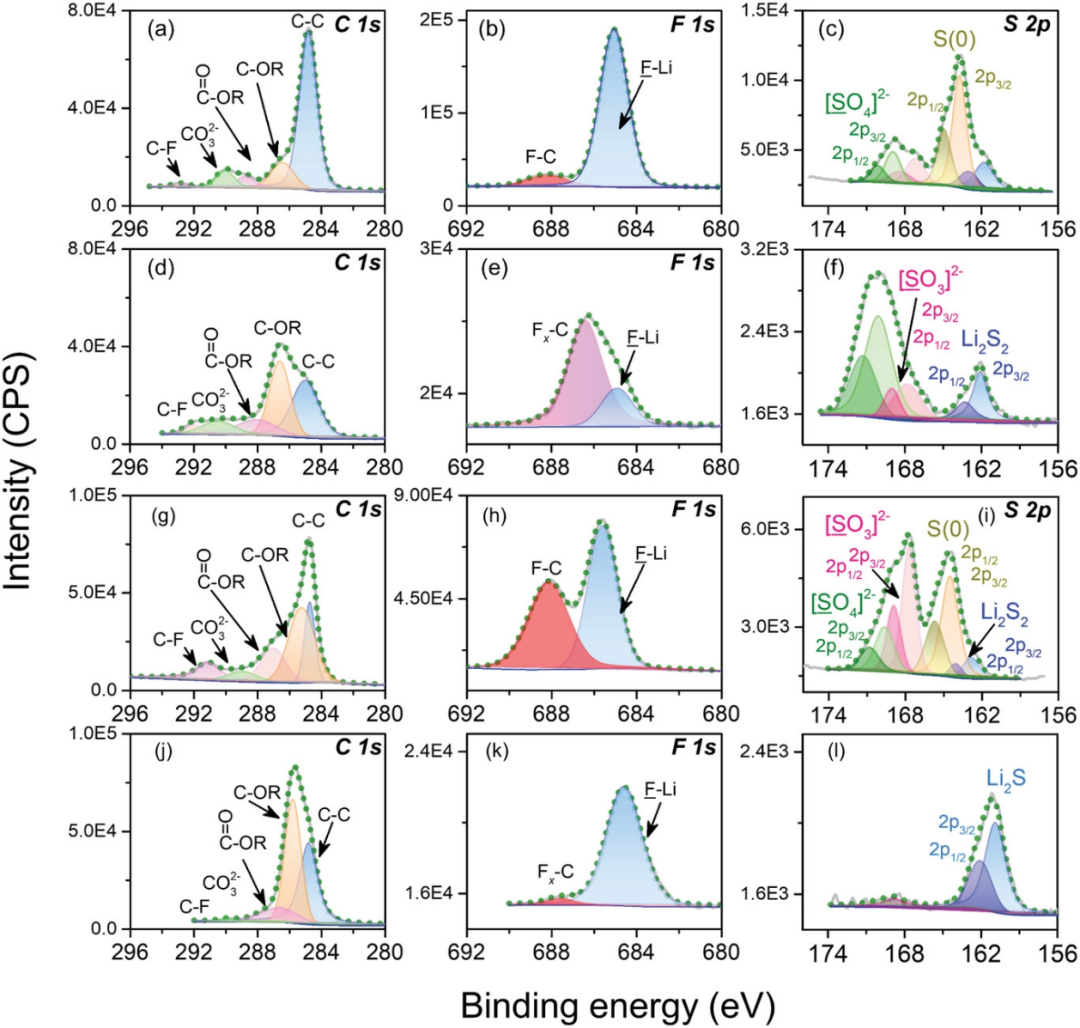
圖10. 兩種電解質中形成的SEI和CEI層的界面表征。500次循環后,a–c)循環負極和g–i)正極與0.1 M電解液的C 1s、F 1s和S 2p的XPS光譜;500次循環后,d–F)循環負極和j–l)正極的C 1s、F 1s和S 2p的XPS光譜,以及1.0 M電解液。
對于正極側,在兩種電解液中循環的電極上其表面膜呈現出更具彈性的碳酸烷基酯和聚二氧戊環(polyDOL)的多孔低聚物,如圖10g,j所示。LCE中的LiF比例要多于標準體系中的LiF,且有相對較高的C-F組分(圖10h,k)。這可能源于具有更多活性位點的硫正極,這是由于0.1 M電解液具有更好的潤濕性能。表面層中硫的形式在S 2p光譜中確定(圖10i,l)。首先,含S物種的信號在SCE中似乎較弱。通過硫物種峰值強度的比較,可以合理推測在SCE中正極上較厚的層,因為含碳化合物較多的較厚層會阻礙硫化合物的檢測信號。通過溶劑分解的有機成分越多(如C-OR,圖10j)將覆蓋電極表面并使原始平滑度變粗糙。更致命的是,在S 2p中幾乎完全存在絕緣硫化物(Li2S,S 2p1/2為160.8 eV)(圖10l)會鈍化硫電極的活性表面,導致容量衰減。相反,LCE中形成的表面層由更多的硫基無機物種組成,如Li2SOx、C/S-S、Li2Sx,它們來自多硫化物的還原(圖10i中的Li2Sx)和完全充電后的氧化產物(圖10i),構成穩定的界面層。因此,低濃度電解液中形成的CEI也是一個混合層,含有更多有機物和一些無機物質,如LiF、Li2Sx和Li3N(圖S10a-c,支持信息),如我們前面所分析的。該混合層可以通過更柔性的界面層更好地適應硫轉化反應的體積變化。此外,在這些無機組分的晶界處可以提供更快速的離子通道。在1 M電解液中,醇基鋰(ROLi)含量明顯較高(圖S10d-f,支持信息)。據報道,它是醚類電解液的主要分解產物。基于上述XPS結果,這可能是解釋0.1 M電解液中更好低溫性能的更合理機制。
【結論】
總之,通過使用低濃度電解液,在低溫環境中實現了具有快速硫轉化動力學、穩定循環和界面化學優化的高性能鋰硫電池。詳細的實驗結果表明,低濃度電解液可以實現快速的轉化反應動力學,特別是對于簡單碳基硫正極中具有挑戰性的固-固反應過程。盡管是基于采用商業大塊硫顆粒(G–S)的正極,使用0.1 M電解液的電池仍舊可以實現900(0.1 C)、750(0.2 C)和600(0.5 C)mAh g–1的放電比容量,與常規1 M電解液相比,并顯示出更小的電壓滯后和更長的第二放電平臺。對界面化學的進一步分析證明,通過自建的低濃度效應,在負極和正極上都存在具有多種成分的更佳保護層。原位電化學阻抗測試進一步證明,在循環過程中,0.1 M電解液中的SEI層電阻和電荷轉移電阻較小,并且短鏈多硫化物(Li2S2)在0.1 M電解液中的溶解度較小,實現多硫化物的穿梭受到抑制。因此,盡管在低溫環境下運行,鋰硫電池仍具有更長的循環耐久性和更好的可逆容量。即使在更低的溫度下(?20℃),在0.2 C下仍可實現600 mAh g–1的高比容量,并保持穩定的容量保持率。考慮到這些積極影響和利用低濃度電解液可有效降低應用成本,這項工作有助于實現在低溫條件下更實用的具有優異性能的鋰硫電池。此外,低濃度電解液可以同時實現高載量硫正極和低電解液體積。因此,確定低濃度電解液中的合理成分尤其重要,這可以確保電池性能和電解液成本得到更好協調。未來的策略,如選擇功能性添加劑和采用優化的共溶劑等,也是很好的方法。
Chu, F., Wang, M., Liu, J., Guan, Z., Yu, H., Liu, B., Wu, F., Low Concentration Electrolyte Enabling Cryogenic Lithium–Sulfur Batteries.?Adv. Funct. Mater.?2022, 2205393.?
https://doi.org/10.1002/adfm.202205393
作者簡介
吳飛翔,教授,博士生導師, 國家海外高層次人才(青年),德國洪堡學者,湖南省杰青,頂尖期刊Materials Today(影響因子26.94)副主編 。中南大學冶金工程學士,中南大學和美國佐治亞理工學院(Georgia Institute of Technology)聯合培養博士。美國佐治亞理工學院(Georgia Institute of Technology)Gleb Yushin教授研究組博士后研究員,德國馬普固體研究所(Max Planck Institute for Solid State Research) Joachim Maier 教授研究組研究員。目前主持海外高層次人才計劃項目、國家自然科學基金青年項目、國家自然科學基金面上項目、德國洪堡基金項目、湖南省杰出青年基金、湖南省重點研發計劃、中南大學特聘教授計劃、中南大學創新驅動等項目。長期開展材料化冶金、高比能二次電池關鍵材料設計與材料界面科學等研究,以第一作者/通訊作者在Advanced Materials, Nano Letters, Energy & Environmental Science , Chemical Society Reviews, Joule, Advanced Functional Materials, Advanced Energy Materials, ACS Nano, Materials Today, Nano Energy等國際頂級期刊上發表學術論文近五十篇。授權中國發明專利7項和國際發明專利2項。
柳斌,湖南大學土木學院副教授,博士生導師,岳麓學者,以第一作者/通訊作者在Advanced Functional Materials,ES&T,Water Researh等雜志發表論文30余篇。
褚福路,山東濟寧人,2015年獲得湘潭大學材料科學與工程學士學位,2018年獲得湘潭大學材料科學與工程碩士學位,碩士期間主要是在中國科學院上海硅酸鹽研究所進行聯合培養(導師 李馳麟研究員),2019年進入中南大學冶金與環境學院攻讀博士學位(新能源材料與器件專業),主要研究方向為高性能鋰金屬負極構筑與電解液體系優化。作為第一作者或共同一作在Adv. Funct. Mater.,ACS Nano, ACS Appl. Mater. Interfaces, J. Energy Chem. 等國際頂級期刊上發表6篇Q1 SCI論文,總計參與并發表了包括Materials Today, Nano Letter, InfoMat在內的15篇SCI期刊論文。
王萌,河南南陽人,2019年畢業于鄭州大學,2022年獲湖南大學碩士學位(導師為柳斌副教授),碩士期間主要在中南大學冶金與環境學院吳飛翔教授課題組進行聯合培養,研究方向為低濃度電解液用于高低溫鋰硫電池。作為共同一作分別在Advanced Functional Materials和Energy&Environmental Materials期刊上各發表一篇SCI論文。
編輯:黃飛
?










 工商網監
工商網監
評論