紙電池:能源儲存新兵
A4紙大的動態電子報紙、自動計價的超市、會“說話”的包裝盒……幾年后,一張0. 5毫米厚的紙電池將創
2010-02-05 09:10:56 1066
1066 一種解決傳統電解質不穩定的方案是使用熔鹽電解質,有研究表明,Li-O2電池可以在熔融硝酸鹽(LiNO3/KNO3)中循環,并具有可逆雙電子氧還原,僅具有0.1 V的電壓滯后。
2022-08-22 10:42:242319 淺談公共機房樣機制作及日常維護隨著電腦和網絡的應用日益普及, 電腦在日常生活中扮演著越來越重要的角色,而學校的教育,無論是高校還是中小學,公共機房都是一個不可缺少的教學實踐基礎硬件設備。而如何高效
2009-10-10 15:04:47
),都是在把電網的電能傳輸到汽車的電池上進行儲存,因為電池只能儲存直流電,所以一般交流電是不能直接到電池的,一般都得通過車載充電機進行(OBC)AC/DC的轉換;
2021-04-19 17:19:55
使用 ADS7142 SAR ADC 和 TLV521 運算放大器實現低功耗系統監控單通道負載開關,可連接/斷開系統負載支持鋰亞硫酰氯 (LiSOCl2) 和鋰二氧化錳 (LiMnO2) 電池系統運行狀況監控待機功耗和電池監控待機功耗均低至 1uA
2018-12-28 15:11:47
樓主翻閱新聞的時候發現電池續航能力好像又有新的進展了,而且可儲存鋰電池五倍電量。我們一起看看是什么樣的技術能做到如此吧。 《自然》上發表了一項研究,描述了一種“鋰氧電池”的原型,其基于超氧化鋰
2016-01-29 14:38:28
鋰二氧化錳電池的反應機理不同于一般電池,在非水有機溶劑中,負極鋰溶解下的鋰離子通過電解質遷移進入到MnO2的晶格中,生成MnO2(Li+)。Mn由+4價還原為+3價,其晶體結構不發生變化。
2020-03-10 09:00:32
以下關于鋰電池各蓄電池的標準內容是我司工程師花了一兩小時整理出來的,優耐檢測帶大家來了解一下:1高空模擬 鋰原電池和蓄電池在運輸中的安全要求 GB 21966-2008 IEC 62281:2016
2020-12-25 15:29:13
能技術完全不同,擊敗鋰離子電池的潛力極大。這種電池的用金屬鋰做負極,在正極一端直接與空氣中的氧氣反應。由于反應物之一是空氣,理論上講,該電池儲存同樣能量所需材料僅為其他電池的一半,其重量也可減半。這一
2018-10-09 10:28:23
還缺乏對何種電極材料能夠促進電池內部電化學反應發生的理解。Shao-Horn和其團隊成員在4月1日出版的《電化學與固態快報》上報道了其研究成果,在鋰-空氣電池中使用金或鉑金電極作為催化劑具有比單一碳
2016-01-13 16:04:23
鋰鐵電池的內部結構如圖1所示。左邊是橄欖石結構的LiFePO4作為電池的正極,由鋁箔與電池正極連接,中間是聚合物的隔膜,它把正極與負極隔開,但鋰離子Li+可以通過而電子 e-不能通過,右邊是由碳(石墨)組成的電池負極,由銅箔與電池的負極連接。
2019-09-30 09:10:42
鋰錳電池在生產過程中使用了低沸點的有機物溶劑,其中有一種叫乙二醇二甲醚(DME)的物質,其閃點溫度較低。在充電過程中,如果電池密封不好,電池發熱造成該物質的揮發,遇到電火花將有可能發生燃燒,產生危險。
2019-11-06 09:10:46
Android系統固件更新機制設計說明文檔V1.1xxx2014-9-14修改歷史記錄內容編制\日期審核\日期批準\日期 V1.0建立初稿Xxx2014-9-14 V1.1 增加配圖,統一英文單詞大小寫Android啟動過程錯誤修正,紅色字體部...
2021-12-20 08:08:24
一種新穎的環路內去塊效應濾波器設計,設計中采用5階流水線的去塊效應模塊,利用混合濾波順序與打亂的存儲更新機制的方法提高了流水線暢順性,濾波一個16×16大小的宏塊僅需要198個時鐘周期。
2021-04-12 06:35:37
LED光源怕硫,這是因為含硫的氣體會通過其多孔性結構的硅膠或支架縫隙,與光源鍍銀層發生硫化反應。LED光源出現硫化反應后,產品功能區會黑化,光通量會逐漸下降,色溫出現明顯漂移;硫化后的硫化銀隨溫度
2014-09-12 09:52:15
一般說明
PL5353A產品是鋰硫離子/聚合物電池保護的高集成解決方案。
PL5353A包含先進的功率MOSFET,高精度電壓檢測電路和延遲電路。
PL5353A被放入超小型SOT23-5封裝中
2023-11-07 10:23:19
PSRAM就是偽SRAM,內部的內存顆粒跟SDRAM的顆粒比較相似,但外部的接口跟SDRAM不同,不需要SDRAM那樣復雜的控制器和刷新機制,PSRAM的接口跟SRAM的接口是一樣的。psram內部
2020-12-28 06:43:38
1.請教一下,像TC275HSM 能支持SecOC中的密鑰刷新機制嗎?即SecOC中的密鑰生成節點生成新的密鑰后,是如何下發給ECU的HSM中?2. 二代HSM TC3xx 的HSM 集成
2024-02-21 06:07:23
范圍-55oC至+85oC鋰金屬含量約 2.5克U.L. 組件識別,MH 121933.6 V伯亞硫酰氯鋰(Li-SOCl2)長期存儲和/或使用后,電壓恢復很快高能量密度低自放電率梭芯結構密封玻璃到
2020-11-18 11:28:42
要的。
幸運的是,Oculus和Crytek一起在VR移動機制的探索上花費了不少時間,現在他們也將成果開放給大家。
在博客中,作者強調開發者應當選取正確的移動機制,或者一系列移動方式的結合,來
2017-08-31 09:17:12
深圳市尊信電子技術有限公司 唐名江 *** WX:tmj8213XB3306A產品是鋰/聚合物電池保護的高集成度解決方案。XB3306A包含先進的功率MOSFET,高精度電壓檢測電路和延遲電路
2021-04-07 14:03:02
的TTL電平定時器和終端啟用(TTE)輸入終止。設備信息功能框圖功能描述bqTINY? 支持適用于單電池的精密鋰離子鋰極化充電系統。圖2顯示了典型的電荷分布、應用電路,圖5顯示了操作流程圖。溫度鑒定注意
2020-09-21 18:17:03
LED光源怕硫,這是因為含硫的氣體會通過其多孔性結構的硅膠或支架縫隙,與光源鍍銀層發生硫化反應。LED光源出現硫化反應后,產品功能區會黑化,光通量會逐漸下降,色溫出現明顯漂移;硫化后的硫化銀隨溫度
2019-07-09 10:26:51
` 檢測煤炭含硫儀器 酒泉煤炭測硫儀,檢測煤炭含硫儀器 酒泉煤炭測硫儀由【鶴壁化驗煤炭含硫儀器】提供的檢測煤炭含硫儀器 酒泉煤炭測硫儀測硫機:***化驗機0392-2121168煤炭快速測硫儀
2019-03-21 20:39:10
鋰鐵電池是近幾年才興起的新一代干電池,其具有容量大、體積小、重量輕、大電流放電、低溫性能優異的特點。相較于堿性電池而言,鋰鐵電池不易漏液,因為電解液完全吸附在隔膜上,電池內部不存在流動性液體,不會
2018-11-27 13:21:49
%。自放電率低(年自放電率≤1%)、儲存壽命長達10年以上。 以1#(尺寸代碼D)鎳鎘電池與1#鋰-亞硫酰氯電池的比能量作一個比較:1#鎳鎘電池的額定電壓為1 2V,容量為5000mAh;1#鋰-亞硫
2014-12-03 18:05:58
(ZnCl2)、鋅二氧化錳(ZnMnO2,即堿性電池)。 還有一系列的鋰電池:鋰-二硫化鐵(LiFeS2)、鋰-二氧化錳(LiMnO2)、亞硫酰氯鋰(LiSOCl2)、氟化碳鋰聚合物(LiCFX)、鋰
2014-08-18 09:33:17
~+70°C (特定產品可低至-60°C)。化學反應式陽極:Li → Li+ + e-陰極:2SO2 +e → S2O4整體:2Li + 2SO2 → Li2S2O4用途雖然鋰二氧化硫電池是以軍事應用為
2014-08-18 10:30:58
在即使是惡劣條件下的排放也十分有限的特性可說是非常優異,可惜的是其電解液具有毒性,并會與水產生反應。此外鋰亞硫酸氯電池具備高能量比、重量輕,但內部阻抗非常高,因此只能以有限的短路電流低速放電;還有一個
2014-08-18 10:20:42
二極管的儲存電荷
2021-03-03 07:10:48
儲能電池模塊(鋰原材料)篇 行業概述隨著城市建設中能源危機和環境污染問題,2021年結合我國實施執行碳中和的遠大目標和大環境下,實現風光互補發電系統多行業應用推行,具備良好超前的發展前景空間
2022-03-11 15:59:46
一次鋰電池(鋰亞硫酰氯電池,鋰硫酰氯電池)的主要市場用途: 1) 檢測儀表: 熱量計;自動儀表讀數器(AMR)---如水表、氣表或電表等;汽車試驗場檢測儀;地震測量儀;石油鉆探檢測儀器;資料記錄器
2015-01-26 10:13:21
一次鋰電池(鋰亞硫酰氯電池,鋰硫酰氯電池)的主要市場用途: 1) 檢測儀表: 熱量計;自動儀表讀數器(AMR)---如水表、氣表或電表等;汽車試驗場檢測儀;地震測量儀;石油鉆探檢測儀器;資料記錄器
2015-01-26 10:23:56
一次鋰電池(鋰亞硫酰氯電池,鋰硫酰氯電池)的主要市場用途: 1) 檢測儀表: 熱量計;自動儀表讀數器(AMR)---如水表、氣表或電表等;汽車試驗場檢測儀;地震測量儀;石油鉆探檢測儀器;資料記錄器
2015-01-26 10:28:54
一次鋰電池(鋰亞硫酰氯電池,鋰硫酰氯電池)的主要市場用途: 1) 檢測儀表: 熱量計;自動儀表讀數器(AMR)---如水表、氣表或電表等;汽車試驗場檢測儀;地震測量儀;石油鉆探檢測儀器;資料記錄器
2015-01-26 10:30:38
一次鋰電池(鋰亞硫酰氯電池,鋰硫酰氯電池)的主要市場用途: 1) 檢測儀表: 熱量計;自動儀表讀數器(AMR)---如水表、氣表或電表等;汽車試驗場檢測儀;地震測量儀;石油鉆探檢測儀器;資料記錄器
2015-01-26 10:33:46
一次鋰電池(鋰亞硫酰氯電池,鋰硫酰氯電池)的主要市場用途: 1) 檢測儀表: 熱量計;自動儀表讀數器(AMR)---如水表、氣表或電表等;汽車試驗場檢測儀;地震測量儀;石油鉆探檢測儀器;資料記錄器
2015-02-02 13:01:23
,積極探索管理新機制。街道納管轄區45家電動自行車生產、銷售、維修單位,并化繁為簡,采取“疏堵結合、以疏為主、***搭臺、企業運作、智慧管理”模式,以電動車保有量為413臺的沙塘布社區為實施試點,計劃建設6個
2018-08-29 14:51:42
顆粒,在正極側接觸,這是難度非常大的。從大家預期的優點上,如果使用了金屬鋰,現在容易燃燒和爆炸的液態電解質,另外使用壽命等等都會延長,模塊配置等都是大家期望的,包括金屬鋰、鋰硫和鋰空氣電池,這些路線在
2017-01-17 09:37:14
和可再生能源過渡的立法,這種短缺將變得更加嚴重。因此,市場迫切需要新的電池技術來解決鋰短缺問題。鋰在元素周期表上的地位幾乎奠定了其能量密度之王的地位。一方面,鋰在可再生能量儲存方面使用壽命成本很低,優勢
2021-04-06 15:03:35
為了研制在電性能、安全性和成本價格等三方面均能較好地滿足電動汽車需求的鋰離子電池,選擇了在氧化鈷鋰中摻雜氧化鎳錳鈷鋰三元材料的方法,研制了新的50Ah動力型鋰離子電池。通過對研制電池進行電性能
2011-03-04 14:30:54
,但是將鋰空氣電池用于商業用途至少還需要10年的時間。 鋰空氣電池的基本化學原理十分簡單。放電時,從負極出發的鋰離子在正極與空氣中的氧氣反應,產生一種叫過氧化鋰的固體產物,填充于碳電極的孔隙中。充電
2016-01-11 16:15:06
淺談新能源汽車電源之鋰硫電池利與弊 一直以來,新能源汽車用動力電池能量密度小和造價高的問題一直困擾著國內外新能源汽車制造企業,為了破解這一難題,近來動力電池生產企業紛紛開始研發
2018-07-13 07:54:40
求幫助啊。我現在一臉懵逼。不太懂刷新機制,想做個電子書但是無奈編程太渣。求大牛指點啊
2016-05-11 09:06:10
檢測硫的設備—化驗硫含量的儀器 檢測硫的設備—化驗硫含量的儀器【全自動測硫儀】英特儀器詳詢:138.3923.4904 煤炭測硫儀,高精度全自動定硫儀,微機快速定硫儀,生物質含硫量分析儀,化驗石油焦
2020-12-28 11:05:45
`檢測螢石全硫總硫儀器哪些設備準確 檢測螢石全硫總硫儀器哪些設備準確 英特儀器螢石含硫量測定儀,測試螢石總硫的儀器,化驗螢石全硫含量的設備,測量螢石硫含量的儀器,螢石含硫量分析儀詳詢
2021-04-13 09:00:45
本文提出了一種具有較高穩定性和安全性、基于bootloader的嵌入式軟件自動更新機制。該更新機制同時保存了3個文件,需要較多的Flash存儲空間,但同時降低了維護成本。
2021-04-27 06:33:59
測硫儀部件結構及功能 定硫儀主要包括高溫裂解爐、送樣機構、電解池和攪拌器、空氣凈化系統、控制系統等。
2019-10-23 09:01:03
電力能源的使用,可用的能量的量很小,往往是不可靠的。傳感器節點通常需要某種方式儲存能量暫時使它準備閱讀時必須采取或信息無線發送。選擇之一是提供一種小型可充電電池或儲能電容器。然而,這些存儲機制有其自身
2016-03-02 16:46:48
磷酸鋰鐵電池廣泛應用于電動車、油電混合車、電動自行車、電動工具機、太陽能LED路燈等。
2019-10-22 09:01:28
鋰空氣電池是一種用鋰作陽極,以空氣中的氧氣作為陰極反應物的電池。 放電過程:陽極的鋰釋放電子后成為鋰陽離子(Li+),Li+穿過電解質材料,在陰極與氧氣、以及從外電路流過來的電子結合生成氧化鋰
2016-01-11 16:27:12
結構緊湊的鋰(Li)電池充電器設計方案
2009-03-26 22:02:01
,理論上正極的容量密度是無限的,可加大容量。另外,如果負極使用金屬鋰,理論容量會比鋰離子充電電池提高一位數。但是,為什么鋰-空氣電池至今都未普及?原因是它存在致命缺陷,即固體反應生成物氫氧化鋰(LiOH
2016-01-12 10:51:49
磷酸鐵鋰電池儲能系統 磷酸鐵鋰電池具有工作電壓高、能量密度大、循環壽命長、綠色環保等一系列獨特優點。并且支持無級擴展。組成儲能系統后可進行大規模電能儲存。 磷酸鐵鋰電池儲能系統由磷酸鐵鋰電池
2015-10-20 16:13:21
信號和槽作為Qt的和新機制,在Qt編程中有著非常廣泛的應用。 事實上,我們在Qt開發中,要想妥善的處理各種事件,根本就離不開信號與槽。信號是事件,信號通過信號處理程序來響應。當一個信號被發射時,相應的信號處理程序就會被調用,在處理程序中編寫代碼來使控件響應事件。
2020-11-20 08:03:11
鈉硫電池是一種用于非移動設備如柵極儲能的熔融金屬電池。鈉硫電池由鈉和硫組成,與其他電池相比,具有非常高的能量密度和非常高的充放電效率。鈉硫電池是由一層固體電解質膜構成的電池。鈉硫電池具有很高的能量
2022-04-28 10:53:27
是使用二氧化錳為正極材料、金屬鋰或其合金金屬為負極材料、使用非水電解質溶液的電池。放電反應:Li+MnO2=LiMnO2(2)鋰離子電池:鋰離子電池一般是使用鋰合金金屬氧化物為正極材料、石墨為負極材料、使用非
2018-03-31 14:19:48
鋰電池包的新機遇,回收再利用!我們首先想到的是家用廢電池和廢鉛蓄電池。而不被人所熟知的,鋰動力電池作為新興的儲能形式開始被人們所接受。 雖然鋰電池包結構復雜,規格不一,回收再利用難度比較大。 但
2018-08-16 09:25:07
【鋰知道】鋰電池基本原理解析:充電及放電機制電池充電最重要的就是這三步:第一步:判斷電壓
2021-09-15 06:47:08
濾布的孔隙從凈氣室排出,脈沖電磁閥有規律地控制壓縮空氣濾布表面的粉塵,粉塵脫離濾布墜入料倉。
五、燒結:高溫燒結工段主要反應原理為,磷酸鐵、碳酸鋰和碳源在以氮氣為保護氣的條件下在電加熱輥道窯中反應成磷酸鐵鋰
2023-09-12 13:22:46
很多人會誤以為鋰離子電池就是鋰電池,實際上兩者是有區別的。那么鋰離子電池和鋰電池的區別在哪里呢? 鋰電池的正極材料是二氧化錳或亞硫酰氯,負極是鋰。舉例來講,以前照相機里用的扣式電池就屬于鋰電池
2015-12-28 15:10:38
|Li電池上這些物質的含量較低。這可以描述為多硫化物穿梭的一種形式。在這種機制中,FSI中的一部分硫首先在鋰金屬負極上脫去氧,最終導致低階硫化物,如Li2S和Li2S2。在電池放電期間,這些硫化物轉化
2022-08-30 08:15:15
新型電池、新型能源不停的進步發展,作為老前輩的鋰電池也不甘落后,最近日本又研發出鋰離子電池的最新正極材料-摻錳鈮酸鋰,據說能量密度有望達6倍,我們快來看看這種正極材料到底是什么,為什么這么厲害吧
2016-01-19 14:06:07
的市場需求。在過去幾十年中,研究人員提出了多種新型負極材料,這些材料通常表現出理想的電勢范圍、更高的容量、優異的倍率性能以及長的循環壽命等優勢,但具有初始活性鋰損失較大(ALL)這一不足。因此,在全電池
2021-04-20 16:15:15
鈷酸鋰、鎳酸鋰和錳酸鋰化合物是近年來鋰離子蓄電池最具吸引力的三種正極材料。從比能量、環境污染和價格方面看,錳酸鋰化合物最具前景。重點闡述了尖晶石型錳酸鋰化合物的制備方法,諸如:固相反應法
2011-03-10 12:46:42
提出了一種基于嵌入式系統的遠程程序更新機制,通過一個具體的嵌入式遠程數字監控系統設計方案,分析了該機制的系統結構、實現原理和實現流程,實際的應用測試表明,所
2009-08-26 11:47:41 14
14 ADO_NET數據集更新機制及并發控制策略:本文分析了8I5J (?> 中的更新機制,論述了三種不同的更新邏輯的產生方式及各自特點,提出了并發控制的一些解決方法,及更新邏輯中其他一
2010-01-01 18:48:3112 嵌入式系統自更新機制的設計與應用
隨著嵌入式系統的發展和廣泛應用,必不可少的維護工作變得日益繁重。如移動電話在用戶使用過程中,部
2009-03-29 15:08:02762 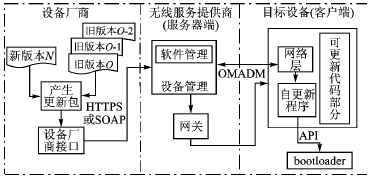
電池儲存期有多久?
就理論上講,電池儲存時總有能量損失。電池本身固有的電化學結構決定了電池容量不可避免地要損失
2009-10-20 13:41:262103 電池儲存應該怎樣存放
根據 IEC 標準規定,電池應在溫度為 20+-5O ° C ,濕度為( 65-+20 ) % 的條件下儲存。一般而言,電池儲存溫度越高,容量的剩
2009-10-20 13:42:404337 電池能儲存多久?
就理論上講,電池儲存時總有能量損失。電池本身固有的電化學結構決定了電池容量不可避免地要損失,主要是由于自放電造成的。通常自放電大
2009-10-28 14:21:56657 Tadiran 電池的儲存與運輸常識
一般性預防措施
2009-10-30 13:07:001569 電池為什么要半電儲存?
2009-10-30 14:26:541452 電池能儲存多久?
就理論上講,電池儲存時總有能量損失。電池本身固有的電化學結構決定了電池容量不可避免
2009-11-13 13:36:403939 氫氧電池反應原理圖
2009-12-18 09:02:58949 文中所提出的新機制采用敲擊鍵盤選擇口令圖片,代替了以往由鼠標點擊的選擇方式,其優點在于可有效地防止肩窺攻擊,并能抵御交叉攻擊,增加了口令機制的安全性。實驗證明,該機制具有
2012-02-13 16:24:2725 全國人大代表、中國科協副主席、中國工程院院士鄧中翰院士表示,將在兩會期間提交三個建議,其中包括“進一步深化科技體制改革、完善自主創新機制”的建議。
2012-03-03 14:56:23642 為滿足航天應用中數據傳輸與存儲中高可靠以及低功耗的要求,實現了一種自適應刷新機制的同步動態隨機存儲(Synchronous Dynamic Random Access MemorySDRAM)控制器
2018-04-03 16:00:440 在美國加利福尼亞州舊金山舉行的IEEE第64屆國際電子設備會議上,三菱電機公司和東京大學提出了提高SiC功率半導體可靠性的新機制。 這個新機制是通過確認柵極氧化物和SiC之間的界面下的硫捕獲器件電流
2019-05-04 12:45:00631 全釩液流電池是將具有不同價態的釩離子溶液分別作為正極和負極的活性物質,分別儲存在各自的電解液儲罐中。在對電池進行充、放電實驗時,電解液通過泵的作用,由外部貯液罐分別循環流經電池的正極室和負極室,并在電極表面發生氧化和還原反應,實現對電池的充放電。
2019-12-02 11:26:1110565 對分層式三維室內地圖分類方法及更新機制說明。
2021-03-17 15:52:510 電子發燒友網為你提供化敵為友,二極管的儲存電荷資料下載的電子資料下載,更有其他相關的電路圖、源代碼、課件教程、中文資料、英文資料、參考設計、用戶指南、解決方案等資料,希望可以幫助到廣大的電子工程師們。
2021-04-06 08:43:564 電容器充電的過程,開關s閉合以后,A極板就通過導線、開關與電源E的負極連接,B極板通過導線與電源正極連接。由于電池正極聚集大量正電荷,就將導線ab中的負電荷吸到a端來,b端就剩下正電荷,在導線b端正電荷作用下,又將B極板中負電荷吸出來,B極板就只剩下正電荷。
2021-06-02 15:36:01918 電池的自放電為什么重要?電池需要多長時間才會自放電? 電池的自放電是指電池在不使用時,由于內部自身的化學反應而自發地失去電荷。這個現象對于電池的使用非常重要,因為它直接影響了電池的儲存能力和使用壽命
2023-11-10 15:01:10523 鋰電池組如何儲存呢? 鋰電池組的儲存是非常重要的,它可以確保電池的長期穩定性和延長壽命。儲存不當可能會導致電池組容量下降、功率衰減,甚至引發安全問題。因此,在儲存鋰電池組之前,有一些關鍵的因素需要
2024-01-10 10:30:29314 高表面積的電極(通常是活性炭)之間的電解質進行電荷的存儲。當電力加到超級電容器時,正負電荷分開存儲在兩個電極上,不涉及化學反應。當電流從超級電容器中流過時,這些儲存的正負電荷被釋放。 1.2 電池的工作原理 電池是通過化學反應
2024-02-04 10:12:31447 電容器接在電路上的作用是什么?會使正負電荷聚集嗎?電壓與電容器并聯會怎樣? 電容器是一種用于儲存電能的器件,它在電路中具有多種重要功能。首先,電容器能夠儲存電荷。當電容器正常工作時,電荷會在電容器
2024-02-06 11:24:00380 ? 堅果派由堅果創建,團隊擁有 12 個華為 HDE,以及若干其他領域的三十余位萬粉博主運營。團隊成員聚集在北京,上海,南京,深圳,廣州,寧夏等地,歡迎合作。 ? 首先鋪墊兩個基礎知識: 1.
2024-03-04 10:02:13145 
 電子發燒友App
電子發燒友App









 工商網監
工商網監
評論