 氟化銅(CuF2)在金屬氟化物正極中的作用
氟化銅(CuF2)在金屬氟化物正極中的作用
01 導讀
由于其高理論電位(3.55 V)和高容量(528 mAh/g),氟化銅(CuF2)在所有金屬氟化物正極中具有最高的能量密度。然而,CuF2只能進行不到5個循環,這主要是由于在充電/放電循環期間Cu離子嚴重溶解。
02 成果背景
近日,馬里蘭大學王春生教授和戴爾豪西大學楊崇英(助理)教授等在電極制造過程中使用水作為漿料溶劑和海藻酸鈉(SA) 作為粘合劑,通過在CuF2顆粒表面形成Cu2+配位的海藻酸鈉(Cu-SA),成功抑制了銅溶解。在0.05 C下循環50次后,具有SA粘合劑的CuF2電極的可逆容量為420.4 mAh g-1,能量密度達到1009.1 Wh kg-1。相關工作以“Super-reversible CuF2 Cathodes Enabled by Cu2+Coordinated Alginate”為題發表在Advanced Materials上。
03 關鍵創新
作者利用在CuF2納米顆粒表面原位形成Cu2+配位SA層的簡單策略,成功地抑制了CuF2正極中Cu離子的溶解,實現了高可逆CuF2正極。
04 核心內容解讀
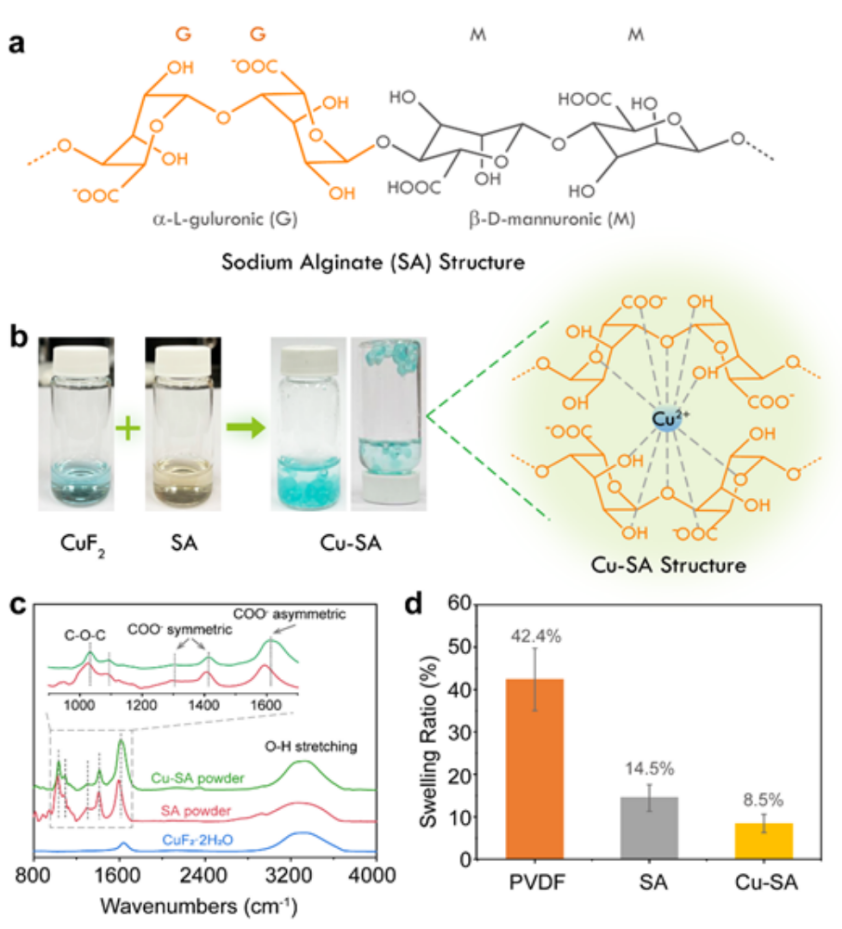
圖1(a)海藻酸鈉(SA)鏈的化學結構;(b)Cu-SA凝膠的照片和Cu-SA的化學結構,直觀地演示了SA和Cu2+離子交聯作用。干燥的SA和Cu-SA粉末的(c)拉曼光譜;(d)PVDF、SA和Cu-SA薄膜在EC/PC溶劑中的膨脹比。@The Authors
作為一種從褐藻中提取的天然多糖,SA包含大量的羥基和羧基(圖1a)。將稀釋的CuF2水溶液滴加入SA水溶液中,SA分子立即與Cu2+離子相互作用,形成一個具有“蛋盒結構”的藍色Cu-SA水凝膠(圖1b)。
作者利用傅里葉變換紅外(FTIR)表征了SA和Cu2+離子之間的強交聯。如圖1c所示,SA粉末在~3300cm-1處表現出氫鍵鍵合的O-H伸縮振動相關的寬吸收帶,在1000~1700cm-1范圍內有一系列對應的尖銳峰。與Cu2+離子交聯后,C-O吸收峰出現明顯的藍移。位于1592、1406和1026 cm-1處的峰與COO-的不對稱/對稱振動和C-O-C基團的不對稱振動有關,分別移至1608、1412和1032 cm-1。這些峰的位移為SA和Cu2+離子之間的化學相互作用提供了強有力的證據。
SA和Cu-SA薄膜在EC/PC有機溶劑中的溶脹性較小,這對有效抑制Cu傳輸和實現快速鋰離子傳輸具有重要意義。如圖1d所示,在EC/PC有機溶劑浸泡24個小時,SA和Cu-SA膜的重量分別增加了14.5%和8.5%,而厚度相近的PVDF膜吸收了大量的碳酸鹽溶劑,重量增加達到42.4%。SA和Cu-SA的低膨脹度,保證了Cu-SA層在CuF2表面的高穩定性。
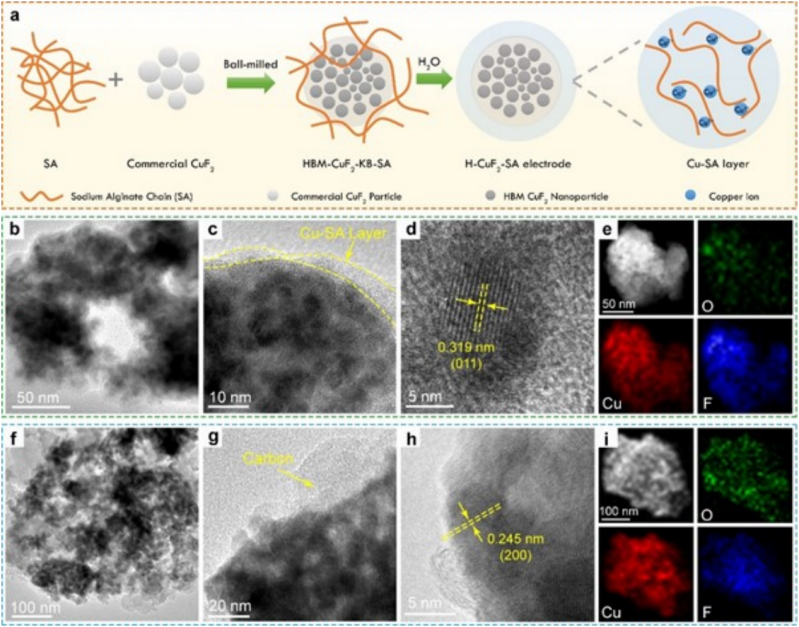
圖2(a)H-CuF2-SA電極的制備示意圖;原始H-CuF2-SA電極納米顆粒的(b)(c)TEM圖像和(d)HRTEM圖像;(e)H-CuF2-SA納米顆粒的HAADF-STEM圖像,及Cu、F、O元素的EDS;H-CuF2-PVDF電極納米顆粒的(f)(g)TEM和(h) HRTEM圖像;(i)H-CuF2-PVDF電極納米顆粒的HAADF-STEM圖像及相應的Cu、F、O元素的EDS。@The Authors
圖2a展示了用SA粘合劑的羥基化CuF2電極(表示為H-CuF2-SA電極)的制備過程。與傳統的電極制備工藝不同,作者沒有直接將SA水溶液與CuF2粒子混合,以避免SA和Cu2+離子之間的快速交聯效應。相反,作者首先用CuF2納米顆粒球磨SA粉,以確保SA的均勻分散,并與CuF2顆粒緊密接觸。然后作者將KB加入球磨的CuF2-SA混合物中,進一步球磨得到CuF2-KB-SA納米顆粒(HBM-CuF2-KB-SA)。
H-CuF2-SA電極粉末的TEM圖像顯示,CuF2表面的Cu-SA層的厚度為~5 nm(圖2b,2c)。從HRTEM圖像中可以看出,Cu-SA層具有無定形結構(圖2c,2d)。Cu、F和O的元素能譜圖顯示了Cu-SA和SA的均勻分布(圖2e)。為了進行比較,作者還采用PVDF粘合劑和NMP溶劑制備了不含Cu-SA涂層的CuF2·2H2O-KB電極(表示為H-CuF2-PVDF電極)。
由于H-CuF2-PVDF中的PVDF粘合劑很難被觀察到,其形態與HBM-H-CuF2-KB納米顆粒幾乎相同(圖2f-g)。平面間距為0.245 nm的單個粒子中的平行晶格條紋與Cu(OH)F的(200)晶格平面有關(圖2h)。同時,HRTEM圖像上的特征圓形晶格條紋是炭黑的特征條紋。
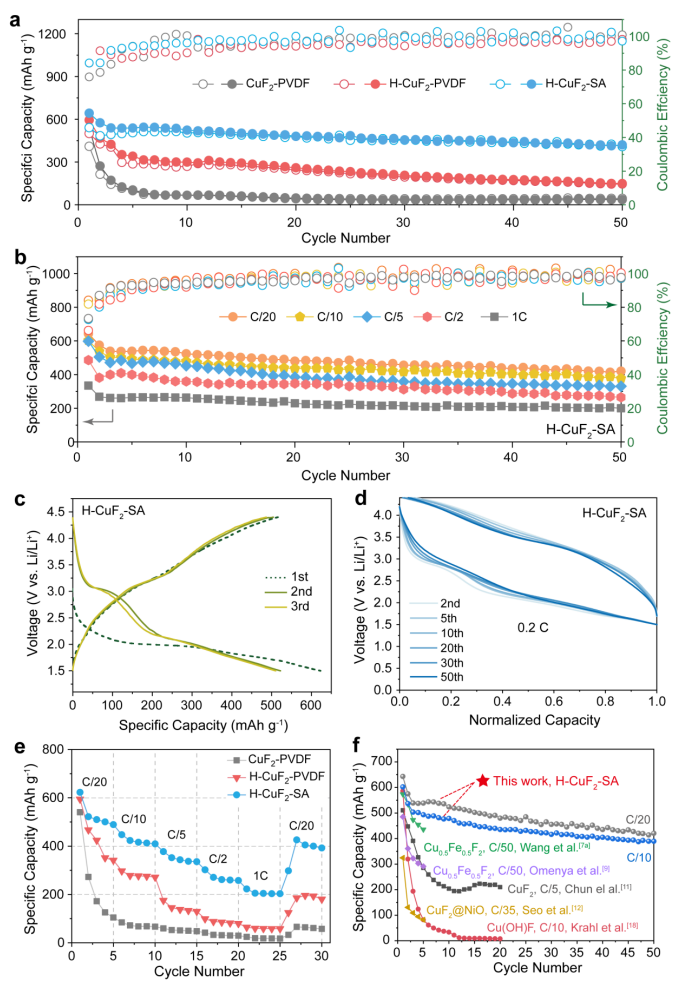
圖3不同粘合劑對無水CuF2和羥基化CuF2正極的電化學性能。(a)在0.05 C下,CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA正極的長循環性能;(b)在不同倍率下,H-CuF2-SA的循環穩定性;(c)H-CuF2-SA在0.05 C(1 C=519 mAh g-1)的充放電曲線;(d)0.2C下,H-CuF2-SA電極的充放電曲線;(e)CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA正極的倍率性能;(f)與其他CuF2或銅基氟化物正極的循環性能比較。@The Authors
圖3a顯示了H-CuF2-SA、H-CuF2-PVDF和CuF2-PVDF電極在0.05 C下的循環性能。僅經過5個循環后,CuF2-PVDF的容量迅速下降到104.5 mAh g-1,低于其初始容量的20%。H-CuF2-PVDF比CuF2-PVDF具有更好的循環穩定性,但在50次循環時,其容量仍下降到148.6 mAh g-1。與之形成鮮明對比的是,H-CuF2-SA在0.05 C下進行50個循環時保持了420.4 mAh g-1的可逆容量,這是以前從未報道過的。
作者還評估了三種電極在不同倍率下的長循環穩定性(圖3b)。H-CuF2-SA(圖3f)在0.05 C時,可達到420.4 mAh g-1的容量,在1 C高倍率下50個循環后保持200.1 mAh g-1,是CuF2-PVDF或H-CuF2-PVDF的5-10倍。與CuF2-PVDF不同,在0.05 C下,H-CuF2-SA和H-CuF2-PVDF的第一次恒流放電曲線顯示出一個在2.0 V左右的長平臺,分別達到622.8和588.3 mAh g-1的高比容量(圖3c)。還原電位較低是由于Cu-OH的電負性低于Cu-F,降低了氟化銅轉化的活化能壘。在第二次及之后的放電中,H-CuF2-SA和H-CuF2-PVDF電極在3.1 V和2.1 V左右均具有較高的放電電位。
由于充電過程中電解質中銅離子的溶解,H-CuF2-PVDF的放電容量迅速衰減。值得一提的是,由于氟化鋰形成的能量勢壘更大,在3.1 V時的容量在初始循環中衰減得更快。由于Cu-SA涂層抑制了Cu陽離子的溶解,H-CuF2-SA顯示出穩定的容量(圖3c)。通過歸一化容量的充放電曲線進一步證明了H-CuF2-SA的可逆轉換(圖3d)。經過50個循環后,H-CuF2-SA在3.0 V和2.1 V左右的還原電位仍清晰可見。
此外,H-CuF2-SA的倍率性能也遠高于H-CuF2-PVDF和CuF2-PVDF(圖3e)。圖3f比較了迄今為止報道的基于CuF2的轉換正極。所有報道的基于CuF2的轉換正極在前5個循環中迅速下降,無論在0.2 C高倍率下或在0.02 C極低的電流密度下。與之形成鮮明對比的是,H-CuF2-SA可以實現50個可逆循環,并保持420.4 mAh g-1的容量,在所有基于CuF2的正極中表現出優越的循環穩定性。
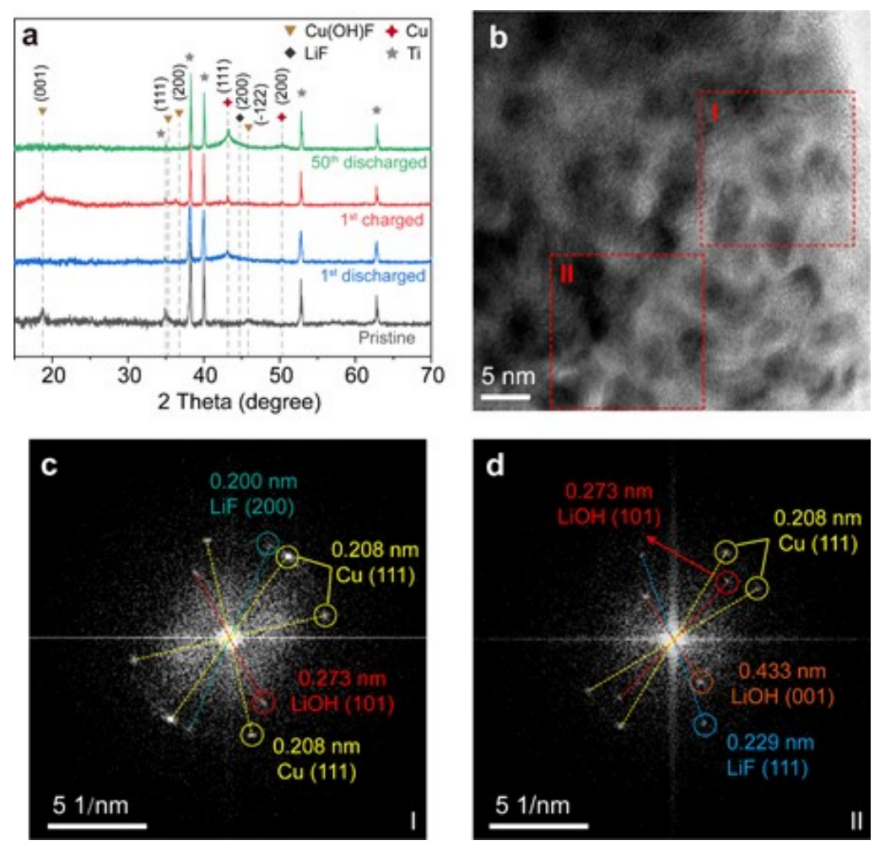
圖4(a)在不同充放電狀態下,H-CuF2-SA正極的XRD;(b)第一次放電后,H-CuF2-SA正極的HRTEM圖像;(c)對應的區域I和(d)區域II的FFT模式。@The Authors
作者通過XRD研究了H-CuF2-SA的相變和反應可逆性(圖4a)。當H-CuF2-SA完全放電到1.5 V時,與Cu(OH)F的(001)平面相關的18.7°處的峰消失,在43.2°左右出現了一個明顯的與Cu的(111)平面對應的寬峰。這表明了Cu(OH)F的完全轉化。作者通過HRTEM圖像和相應的FFT模式,進一步證明了LiF和LiOH的存在(圖4b-d)。由于Cu、LiF和LiOH納米顆粒的疊加分布,它們在HRTEM圖像中的晶格平面存在重疊,難以區分(圖4b)。對HRTEM圖像的兩個選定區域(圖4b中的紅框)進行了快速傅里葉變換(FFT)分析,以闡明LiF和LiOH的存在。
區域I對應的FFT模式顯示了三組衍射點,表明了該區域的多晶分布(圖4c)。晶格平面距離為0.208 nm的最亮衍射點與高結晶Cu的(111)平面有關,這與XRD的結果一致。晶格距離較低的暗點與LiF的(200)平面有關。由于(111)平面與Cu的平面距離最大,因此用Cu可以很容易地分辨出表現出較大平面距離的衍射點。因此,在區域I和區域II,位于較小圓上的衍射點可以根據其特征晶格距離分別對應到LiF的(111)平面、LiOH的(101)和(001)平面(圖4c,d)。
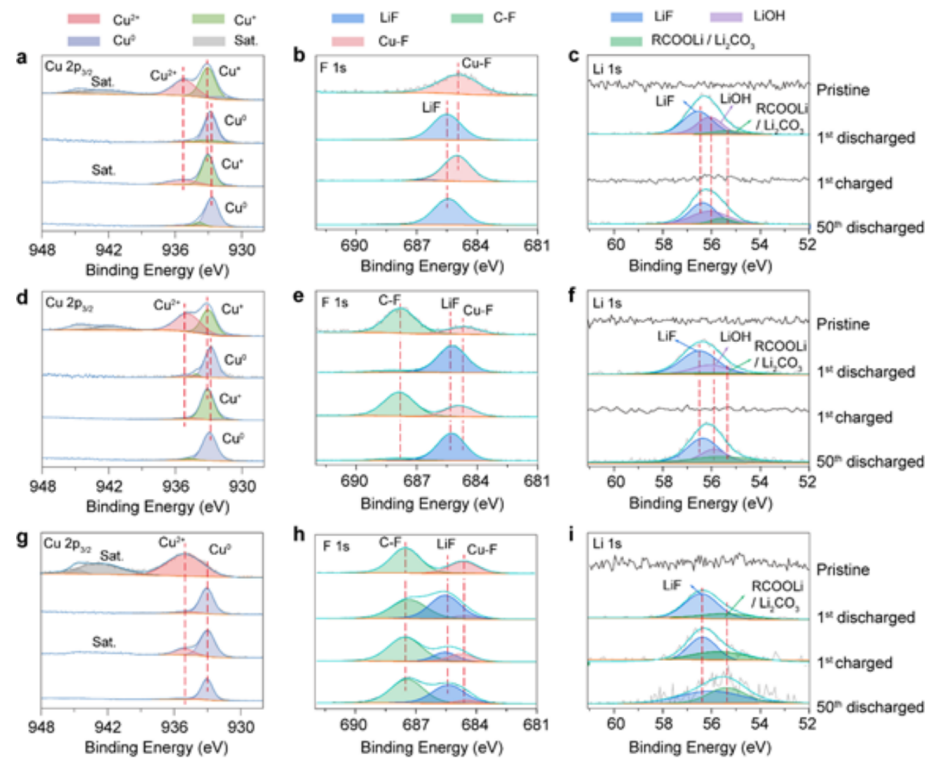
圖5(a-c)H-CuF2-SA;(d-f)H-CuF2-PVDF和(g-i)CuF2-PVDF電極循環前后的Cu 2p2/3、F 1s和Li 1s的XPS光譜。每個光譜都從電極表面進行測試。@The Authors
對原始和循環后的CuF2-PVDF、H-CuF2-PVDF和H-CuF2-SA電極進行XPS分析,以揭示它們的可逆性(圖5)。對于兩個原始的H-CuF2-SA(圖5a)和H-CuF2-PVDF(圖5d)電極,Cu 2p3/2光譜可以很好地模擬為位于933.1 eV和935.2 eV處的兩個峰,這可以分別歸因于Cu+和Cu2+的氧化態,表明了銅在Cu(OH)F中的多價性質。而在原始CuF2-PVDF電極中,只觀察到一個與Cu2+-F相關的在935.7 eV的單峰(圖5g)。
當電池第一次放電至1.5 V,所有三個電極的Cu 2p3/2光譜均移到932.6 eV,對應于金屬Cu0,表明這些氟化物電極完全還原。在第一個充滿電狀態下,原始樣品在H-CuF2-SA電極中恢復了Cu2+和Cu+的價態,具有良好的轉化反應可逆性。而在H-CuF2-PVDF中,由于電極表面充電過程中銅的嚴重溶解,Cu2+信號幾乎消失,這與其充放電性能相一致。對于CuF2-PVDF電極,Cu0峰的存在表明,由于Cu顆粒的粗化,只有部分金屬銅被重新轉化為CuF2。純CuF2的不完全轉換主要是由于導電金屬銅與分離的LiF聚集體之間的電子傳輸中斷,導致CuF2-PVDF電極中存在大量的“死”活性物質。
放電狀態下的Li1s光譜顯示了純CuF2和羥基化CuF2之間不同的轉化反應路徑(圖5c,f,i)。在放電狀態下,H-CuF2-PVDF和H-CuF2-SA電極的F1s光譜中都可以檢測到Li-F和Li-OH,這有力地證明了在還原反應中形成了LiF和LiOH的混合物,而不是純LiF。LiOH的存在有效地降低了轉化反應的自由焓,從而保證了Cu(OH)F的反應比純CuF2正極材料更徹底。
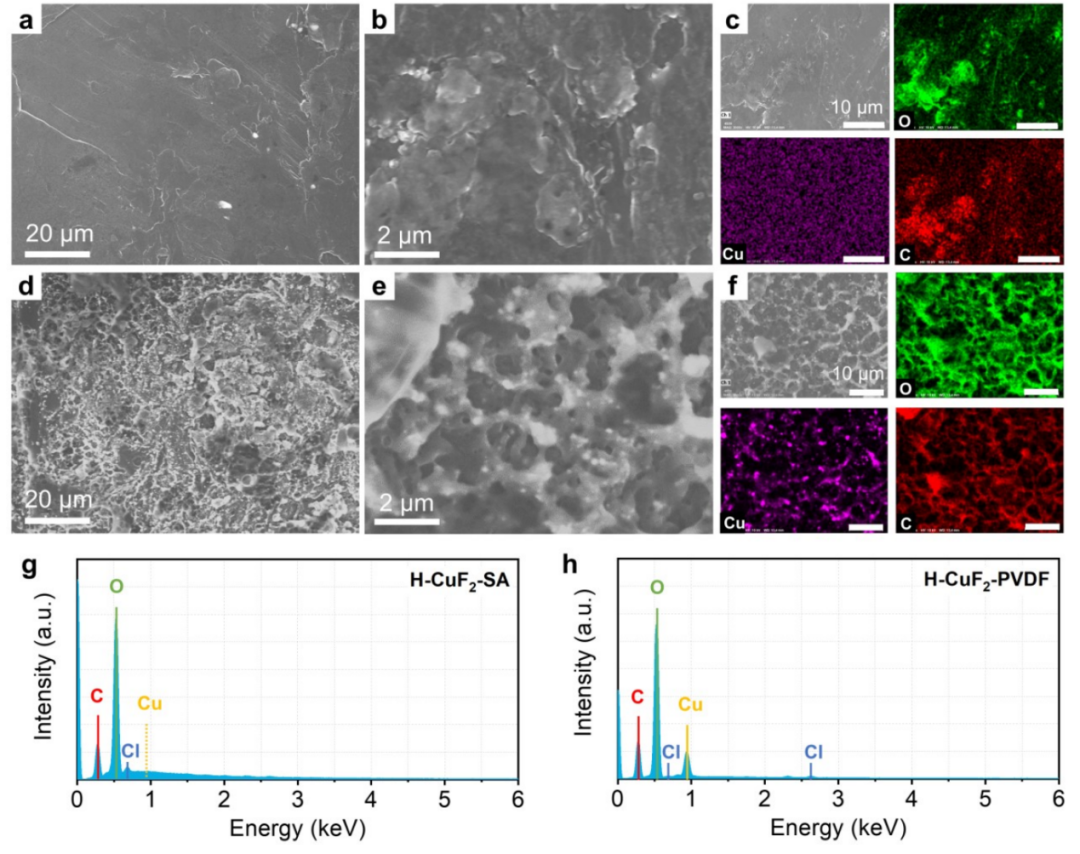
圖650圈循環后,與(a-c)H-CuF2-SA和(D-F)H-CuF2-PVDF配對的Li負極的SEM圖像,及相應的Cu、O、C元素能譜圖。(g)H-CuF2-SA和(h)H-CuF2-PVDF配對的Li負極循環后的表面的EDS。@The Authors
由于銅離子在電解液中的溶解會沉積在鋰負極上,因此可以用掃描電鏡和EDS進行分析。如圖6a-b所示,與H-CuF2-SA正極結合的循環后的Li負極,表面光滑,只有少量鹽殘留可見,圖6c所示的C和O元素圖像進一步證明了這一點。在相應的EDS光譜中沒有觀察到Cu信號的痕跡(圖6g),這表明H-CuF2-SA的鋰化/去鋰化過程中沒有發生明顯的銅溶解。
與之形成鮮明對比的是,與H-CuF2-PVDF正極結合的循環后的Li負極的SEM圖像顯示,表面極其粗糙,大量的Cu粒子被鍍在Li表面(圖6d-e)。Cu元素能譜圖中的亮藍色點和相應EDS光譜中的強Cu峰進一步顯示了Cu的顯著含量(圖6f,6h),表明H-CuF2-PVDF中銅溶解嚴重。
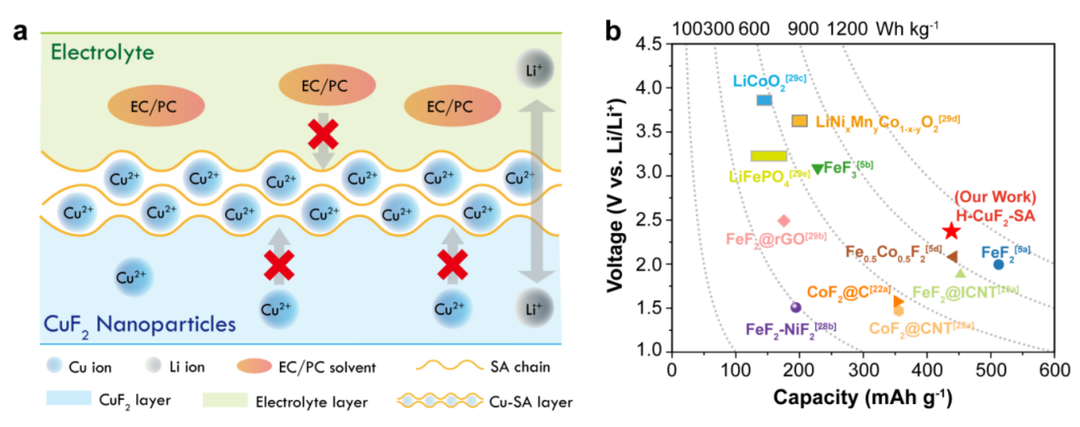
圖7(a)SA粘結劑增強H-CuF2-SA電化學性能的工作機理示意圖;(b)H-CuF2-SA與其他金屬氟化物和插入正極的容量、電壓和能量密度的對比,放電能量密度基于0.05 C的放電電流計算。@The Authors
對H-CuF2-SA的綜合表征和電化學分析表明,H-CuF2-SA的超循環穩定性歸因于在電極制造過程中原位形成的獨特的Cu-SA涂層。圖7a示出了H-CuF2顆粒上Cu-SA涂層的形成和功能。在電極制備過程中,由于SA鏈與水溶液中銅離子之間的強交聯效應,在CuF2顆粒表面原位形成了堅固的Cu-SA層。Cu-SA層對Li+有選擇性的滲透性,而對Cu2+具有不滲透性,有效地抑制了Cu離子的溶解和降解。此外,Cu-SA層和SA粘合劑在碳酸酯電解質溶劑中的膨脹性都較低,降低了電極/電解質的相互作用,防止了電解液進入粘合劑/電極界面,從而進一步降低了銅在電極中溶解的可能性。
雖然,水基SA粘合劑將水合雜質引入到CuF2材料中,但H-CuF2-SA仍能提供2.4 V以上的平均放電電位。高容量與高放電電壓相結合,在50次循環后,電流密度為0.05 C時,可逆放電能量密度為~1009.1 Wh kg-1(圖7b),高于大部分金屬氟化物。更重要的是,本研究中H-CuF2-SA的能量密度高于大多數鐵基氟化物和商用插入正極材料。
05 成果啟示
本文通過在CuF2納米顆粒表面原位形成Cu2+配位SA層,成功地抑制了CuF2正極中Cu離子的溶解。在0.05 C下50次循環后,獲得的H-CuF2-SA電極的可逆容量為420.4 mAh g-1,達到1009.1 Wh kg-1的高能量密度。其優越的循環穩定性和能量密度證明了銅基氟化物正極的最佳性能。
審核編輯:劉清
-
PVDF
+關注
關注
1文章
32瀏覽量
10201 -
TEM
+關注
關注
0文章
85瀏覽量
10393 -
HBM
+關注
關注
0文章
373瀏覽量
14707
原文標題:王春生&楊崇英Adv. Mater.:高可逆氟化銅正極
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
氣體濃度監測師——六氟化硫sf6泄漏報警在線監測系統

配電室六氟化硫SF6設備泄漏監測方法

電力系統為何要進行六氟化硫氣體SF6泄漏在線監測?
電廠配電室為什么要安裝六氟化硫泄漏監測報警系統 ?

浸沒式液冷散熱氟化液新產品 DAISAVE

電力環境六氟化硫氣體泄漏監測解決方案
配電室為什么要安裝六氟化硫泄漏在線報警監測裝置?
六氟化硫氣體泄漏報警裝置,安全保障的重要一環!
TE推出SPEC55低氟化物電線和電纜哪里有?-赫聯電子
SF6環網柜六氟化硫氣體泄漏報警在線監測系統
LW□-252戶外高壓交流六氟化硫斷路器
電力系統為何要進行六氟化硫氣體在線監測?
六氟化硫SF6在線監測系統,氣體泄漏檢測的小幫手





 工商網監
工商網監
評論