 使用基于Mo礦物水凝膠設計不含碳的單個鐵原子分散的異質結構納米片
使用基于Mo礦物水凝膠設計不含碳的單個鐵原子分散的異質結構納米片
研究背景
氫氣因具有能量密度高、在空氣中燃燒時排放的污染物少等優點而成為一種有備受關注的能源。在各種制氫方法中,電化學方法是較容易和更經濟的。其中商業貴金屬基HER電催化劑的高成本促使研究人員開發具有高活性和高穩定性的低成本電催化劑。具有單金屬原子分散體(SACs)的異質結構材料是制氫的理想催化劑材料。然而,大規模制備高穩定性和低成本異質結構錨定單原子的催化劑仍然是一個巨大的挑戰。
成果簡介
鑒于此,香港城市大學呂堅院士、李揚揚,哈爾濱工業大學孫李剛(共同通訊作者)等提出使用基于Mo礦物水凝膠作為前體來設計新的不含碳(C)的單個鐵原子分散的異質結構納米片作為析氫催化劑。Fe/SAs@Mo基HNSs在HER中表現出優異的電催化活性和長期耐久性:在10mA cm-2下的過電位為38.5 mV,Tafel斜率為35.6mV dec-1,在200mA cm-2下可連續運行600 h。
研究亮點
1、通過低溫磷化自組裝無機-無機配位的FePMoG納米片,得到了一種高效的HER電催化劑Fe/SAs@Mo基HNSs;
2、Fe/SAs@Mo基HNSs的異質結構界面和單一分散的鐵原子有利于HER過程中H2O的吸附和適當的H*解吸附。
圖文介紹
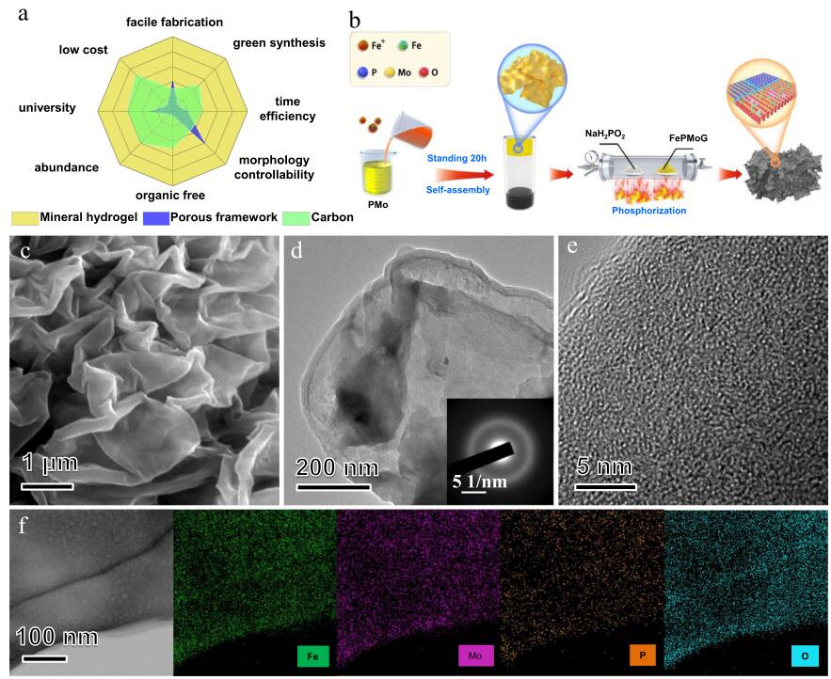
圖1 制備FePMoG和Fe/SAs@Mo基HNSs電催化劑的概念設計和微觀結構表征。
礦物水凝膠由無機物、無機鹽和水制成,可以在溫和條件下通過快速簡單的混合和膠凝過程自組裝成各種形態的凝膠網絡。在加熱處理后,單獨分散在凝膠網絡中的配位金屬離子可以轉化成金屬氧化物/磷化物/硫化物的異質結構。與其他常見的單原子基底前驅體(多孔框架和碳)相比,礦物水凝膠在合成路線、環境友好、生產效率、可調節性、原材料豐富性、可持續性、和成本效益方面顯示出巨大的優勢(圖1a)。圖1b顯示了制備FePMoGs納米片的新型配位誘導自組裝工藝過程。首先在室溫下簡單混合多金屬氧酸(PMo)和三價鐵離子(Fe3+)的溶液。通過配位誘導的自組裝過程,形成了均勻分布的FePMoG納米片的懸浮液。25∶1摩爾比的Fe3+: PMo的混合物可以形成寬度為幾微米,厚度約為50納米,且表面光滑起皺的納米片(圖1c)。圖1d中的FePMoG的TEM圖像證明了FePMoG復合材料的二維結構,且SAED圖案(圖1d中的插圖)為寬衍射環,證實了其無定形性質。HR-TEM圖像(圖1e)顯示了無定形FePMoG中原子的無序排列。FePMoG的STEM圖像和相應的EDS揭示了Fe、Mo、P和O原子在其結構中的均勻分布,表明它由Fe3+和PMo之間的均勻相互作用組成(圖1f)。
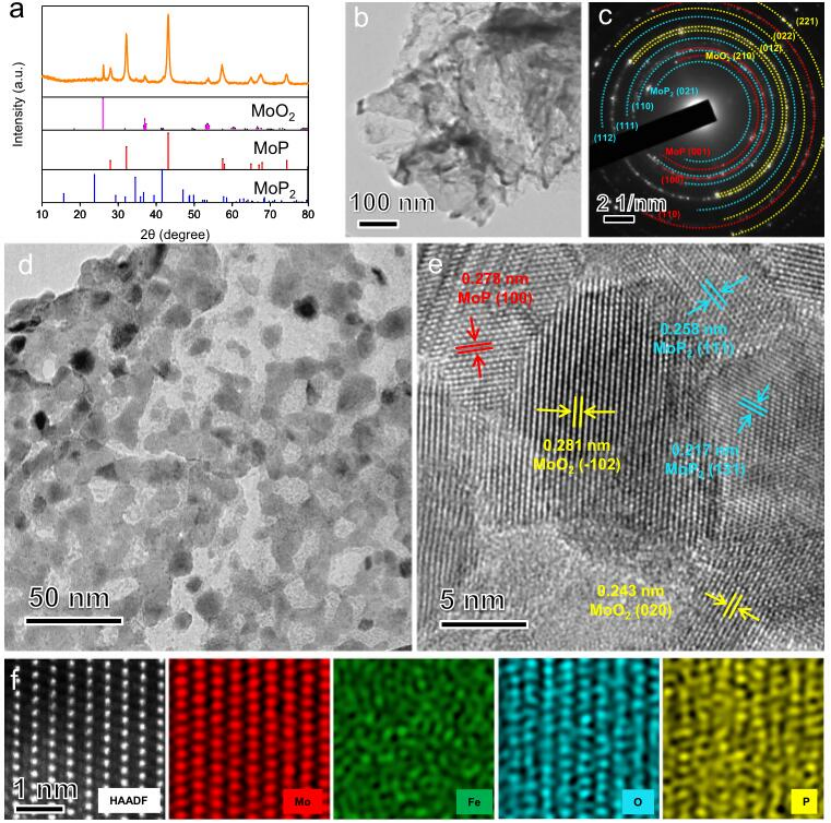
圖2 Fe/SAs@Mo基HNSs的結構表征。
磷化后FePMoG納米片的結構演變如圖2所示。Fe/SAs@Mo基-HNSs的XRD圖案表明存在二氧化鉬(MoO2,JCPDS:78-1069)、磷化鉬(MoP,JCPDS:240771)和二磷化鉬(MoP2,JCPDS:89-2678)的特征衍射峰(圖2a)。Fe/SAs@Mo基HNSs在磷化過程中保持其初始2D多孔納米片形態(圖2b)。相應的SAED圖包含由MoO2,MoP,和 MoP2形成的環(圖2c)。高倍率TEM圖像顯示Fe/SAs@Mo基-HNSs(圖2d)包含平均直徑<10 nm的孔的互連結構。圖2e中的晶格條紋為0.2 81,0.243,0.278,0.209,0.258,和0.217 nm,分別對應于MoO2(102)、MoO2(020)、MoP(100)、MoP(101)、MoP2(111)和MoP2(131)的晶面間距,表明Fe/SAs@Mo基-HNSs的異質結構。Fe/SAs@Mo基-HNSs的HAADF-STEM圖像和相應的EDS元素圖揭示了Fe、Mo、P和O原子的均勻分布,并且Fe原子獨立地分散在晶格中(圖2f)。
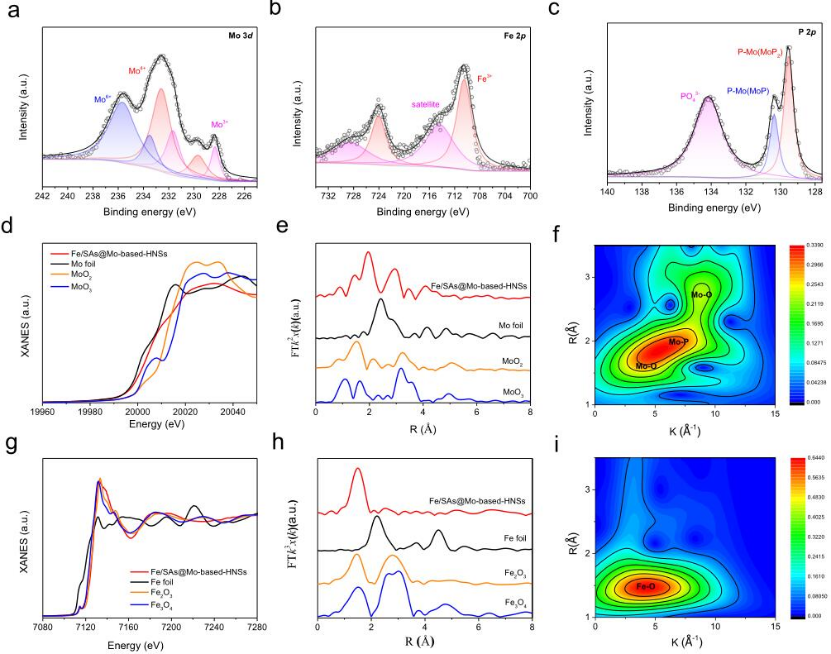
圖3 Fe/SAs@Mo基HNS的XPS和XANES光譜。
采用XPS和XANES表征了Fe/SAs@Mo基-HNSs的化學組成、價態和配位狀態以及電子結構(圖3)。如圖3a所示,Mo 3d的高分辨率XPS光譜由六個峰組成:231.66和228.34 eV (Mo3+)、232.58和229.68 eV(Mo4+)以及235.63和233.49 eV(Mo6+),分別對應于MoP、MoO2和MoP2。Mo K邊緣XANES光譜顯示Fe/SAs@Mo基HNSs的近邊緣吸收能介于Mo箔和MoO2的吸收能之間,表明Mo的平均氧化態介于Mo0和Mo4+之間(圖3d)。相應的EXAFS揭示了分散在Fe/SAs@Mo基HNSs中的Mo原子與P和O原子配位,不存在Mo-Mo鍵(圖3e,f)。Fe的高分辨率XPS光譜(圖3b)在710.7 eV (Fe 2p3/2) 和724.1 eV (Fe 2p1/2) 處具有Fe3+的特征峰,并且在715.6 eV處具有Fe3+2p衛星峰。Fe K邊XANES光譜也顯示Fe的化合價接近Fe3+(圖3g)。相應的EXAFS結果描繪了鐵原子的配位環境:在約1.5埃處的強峰歸因于Fe-O鍵,在傅里葉變換EXAFS中,在2.47或2.85埃處不存在Fe–Fe峰(圖3h),這表明Fe在Fe/SAs@Mo基HNSs中是孤立存在的,并且大部分與O進行配位(圖3i)。
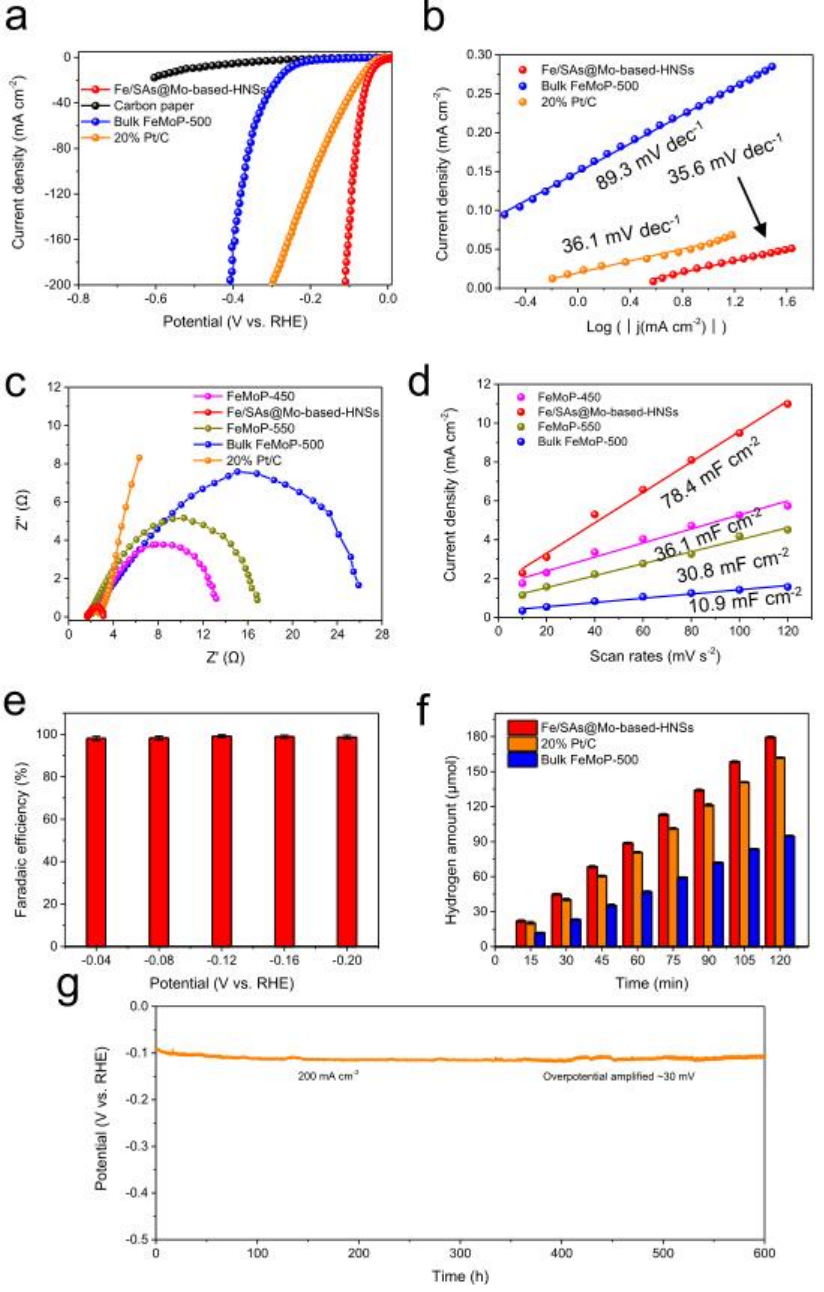
圖4 Fe/SAs@Mo基HNSs在1.0?M KOH溶液中的HER電催化性能。
使用典型的三電極系統,在1 M KOH溶液中,研究了Fe/SAs@Mo基HNSs的HER性能。圖4a所示的線性掃描伏安圖表明Fe/SAs@Mo基-HNSs具有優異的催化性能:在10mA cm-2下,過電位僅38.5 mV,比FeMoP-T樣品和商業20 wt% Pt/C的低得多。當電流密度達到200mA cm-2時,Fe/SAs@Mo基-HNSs的過電位略微增加到109.9 mV,顯著低于商業20 wt% Pt/C的過電位(299.3 mV)。圖4b表明Fe/SAs@Mo基HNSs的Tafel斜率(35.6mV dec-1)小于FeMoP-500(89.3mV dec-1)和其他FeMoP-T樣品,而接近20% Pt/C(36.1mV dec-1)。如圖4c所示,Fe/SAs@Mo基HNSs具有最小的電荷轉移電阻。Fe/SAs@Mo基-HNSs的Cdl為78.4 mF cm-2(圖4d),遠高于直接磷化的塊體FeMoP-500(10.9 mF cm-2)以及FeMoP-450和FeMoP-550樣品的Cdl (分別為36.1和30.8 mF cm-2)。從圖4e中可以看出,Fe/SAs@Mo基HNSs的法拉第效率在寬電勢下接近100%。Fe/SAs@Mo基HNSs的H2產率隨時間線性增加,并且顯著高于商業20 wt% Pt/C和塊狀FeMoP-500的產率(圖4f)。Fe/SAs@Mo基HNSs的穩定性也非常好:200mA cm-2的電流密度下可保持600 h(圖4g)。
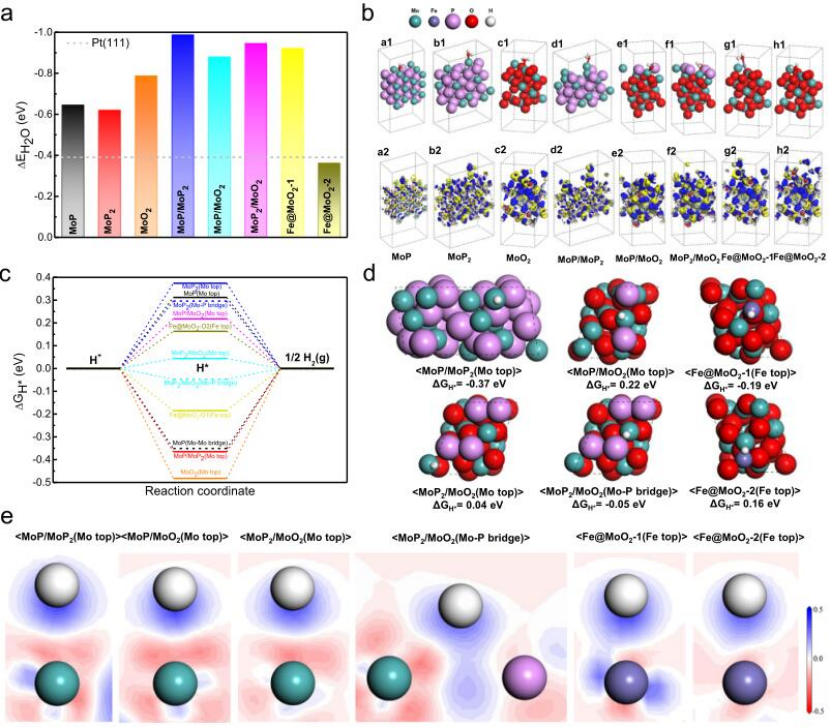
圖5 設計用于在堿性條件下催化HER的Fe/SAs@Mo基HNS的DFT模擬。
采用DFT計算確定了Fe/SAs@Mo基HNSs在HER中的突出電催化活性的分子基礎。計算了水分子在催化位點上的吸附能(ΔEH2O),圖5a表明,所有界面模型都比單相模型具有更高的ΔEH2O值,證明異質結構界面有效地促進了H2O吸附。為了進一步解釋增強H2O分子吸附的原理,DFT模擬的原子構型和在Mo或Fe位點的H2O吸附后相應的電子密度差異如圖5b所示。結果表明異質界面和單個Fe原子位置在吸附H2O分子時表現出明顯的電子損耗,證明有效促進了電子從活性位置轉移到H2O分子。
為了檢驗H*吸附/H2解吸步驟(Heyrovsky步驟),計算了所有可能的活性位點的吉布斯自由能(ΔGH*)(圖5c),相應的原子構型顯示在圖5d中。圖5c顯示,單相模型中所有活性位點上H*吸附的ΔGH*值超出0.3–0.3 eV的范圍,表明HER性能較差。而異質結構界面和單原子分散模型中大多數活性位點的ΔGH*值在0.19–0.22 eV范圍內(MoP/MoP2除外)。特別地,MoP2/MoO2中的
為了闡明H*吸附的物理機制,通過研究H*吸附在異質界面和單原子分散模型的表面位點上產生的二維電子密度差異(圖5e),分析了H*和活性位點之間電子轉移的大小。在Fe單原子分散模型中,發現Fe@MoO2-1中Fe位點的電子轉移比Fe@MoO2-2中的大。這與它們的ΔGH*值的差異相一致(Fe@MoO2-1為-0.19 eV,Fe@MoO2-2為0.16 eV)。有利的ΔGH*值表明,在MoO2中與一個或兩個O原子結合的單原子分散的鐵原子具有適當的H*吸附/解吸能力,可以有效地進行HER電催化。
總結與展望
本文通過對環境友好自組裝有機無機配位的FePMoG納米片進行低溫磷化制備了一種由多孔的Fe/SAs@Mo基HNSs組成的高效HER電催化劑。電化學測試表明此催化劑具有迄今為止報道的納米結構的Mo基電催化劑的最佳HER性能之一。這種出色的性能歸因于Fe/SAs@Mo基HNSs優化的電子結構、豐富的界面和邊界相、較大的活性表面積和孔隙率,以及單原子和異質結構的協同效應。這種高性能的無碳電催化劑是易腐蝕的含碳催化劑的有效替代品,可用于質子交換膜燃料電池和其他先進的能源技術。
審核編輯:郭婷
-
納米
+關注
關注
2文章
693瀏覽量
36953 -
能源
+關注
關注
3文章
1577瀏覽量
43424
原文標題:Nature Commun.:二維礦物水凝膠衍生的異質結構錨定單原子,實現超穩定析氫反應
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
水凝膠半導體材料問世,有望用于生物集成電路
探索 32KHz MEMS 振蕩器的卓越性能:μPower MO1532、MO1552、MO1630、MO1566 和 MO1568

MEMS 振蕩器:Low Jitter MO9120 MO9121 MO9122 MO8208 MO8209 的卓越性能與應用
具有密集交聯結構的明膠基水凝膠電解質(ODGelMA)
通過2D/3D異質結構精確控制鐵電材料弛豫時間

九號電動將于4月19日發布新品智能碳晶電池
基于顏色變化水凝膠的集成微流控壓力傳感

什么是無定形碳膜?為什么選碳作為3D NAND的硬掩模?

無分散劑膠體與硅-納米碳界面工程制備高性能鋰離子電池負極材料





 工商網監
工商網監
評論