 一文簡析磷表面氧化提高儲鋰性能
一文簡析磷表面氧化提高儲鋰性能
01
研究背景
由于具有較高的理論容量(2596 mAh g–1)和適當的鋰化電勢(~0.7 V vs Li+/Li),磷被認為是鋰離子電池最有前途的負極材料之一。然而,它具有大的體積變化(~300%),低電導率(~10–14S cm–1)和不穩定的固體電解質界面(SEI)等缺點,導致電化學循環衰減迅速。
為此,研究人員采用了各種策略來解決這些問題,包括結合各種碳框架來實現P顆粒的納米限制,引入導電聚合物涂層以緩解體積膨脹,以及應用功能電解質來構建高彈性SEI膜。盡管上述問題已經基本解決,但磷在制備過程中容易被氧化。因此,磷基負極材料的粉末合成、漿料制備和電極烘焙對避免氧化有很高的要求。
02
成果簡介
近日,天津大學孫潔教授和貴州振華電子化工有限公司Qianxin Xiang在Nano Letters上發表了題為“Surficial Oxidation of Phosphorus for Strengthening Interface Interaction and Enhancing Lithium-Storage Performance”的論文。該論文展示了一種原位預氧化策略,以在磷顆粒上構建功能性氧化層。氧化層不僅作為保護層延長了磷負極在空氣中的儲存時間,還碳化了N-甲基吡咯烷酮和聚偏氟乙烯,加強了磷顆粒與粘結劑之間的界面相互作用。氧化物層進一步誘導形成具有高鋰離子電導率的穩定固體電解質界面。在100次循環后, 氧化的P-CNT保持1306 mAh g–1的高比容量,容量保持率高達89%,遠高于原始P-CNT(17.1%)。原位氧化策略簡單易行,有利于磷基負極的實際應用。
03
研究亮點
(1)在漿料制備過程中,氧化磷可以碳化N-甲基吡咯烷酮(NMP)溶劑和聚偏氟乙烯(PVDF)粘結劑,加強磷顆粒與粘結劑之間的界面相互作用。
(2)氧化層可以作為保護層,延長磷負極在空氣中的儲存時間。
(3)氧化物層可以進一步誘導形成具有高鋰離子電導率的穩定SEI。
04
圖文導讀
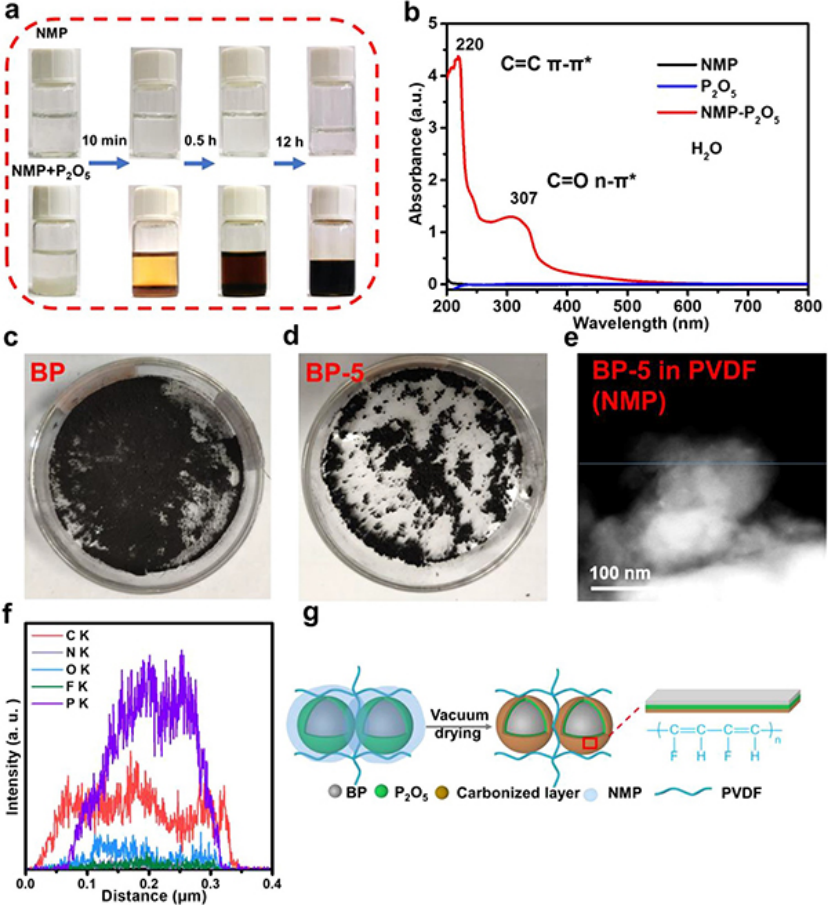
圖 1、(a)分別在真空干燥10分鐘、0.5小時和12小時后,僅NMP溶劑和含有P2O5的NMP的光學圖像。(b)含有P2O5的NMP,僅含有NMP,僅含P2O5紫外可見光譜。(c)原始BP粉末和(d)氧化BP粉末圖像。(e-f)BP-5在含有3%PVDF的NMP中的STEM圖像和線掃描結果。(g)漿料制備過程中碳化層形成的示意圖。
在漿料制備過程中,NMP和PVDF粘結劑與磷氧化物發生碳化反應,因為磷氧化物是強脫水和碳化劑,而NMP和PVF是含有碳、氫、氮和氟的有機物質。為了驗證這一點,將P2O5添加到NMP中,將其置于80°C的真空干燥箱中。當P2O5在10分鐘后添加到NMP中時,溶液的顏色呈現為淺黃色(圖1a)。當時間分別延長到0.5小時和12小時時,溶液的顏色變得越來越深。然而,即使在相同條件下12小時后,純NMP溶劑的顏色仍保持無色和透明。
將反應12小時的純NMP和NMP+P2O5溶液干燥以完全去除NMP,并分別在等體積的去離子水中重新溶解,以進行UV-vis光譜測試(圖1b)。NMP+P2O5的紫外-可見光譜分別在220和307 nm附近顯示出兩個不同的吸收峰(圖1b)。220 nm附近的吸收峰歸因于C═C鍵的π–π*電子能級躍遷,而307 nm處的吸收峰歸因于C═O鍵的n–π*躍遷。
為了消除殘余P2O5和NMP的干擾,將P2O5粉末作為對照樣品添加到去離子水中,以進行UV-vis光譜測試。P2O5(藍線)和NMP(黑線)的對照樣品在220和307 nm處沒有出現類似的吸收峰(圖2b),表明C═C鍵來源于NMP的碳化反應。
為了進一步驗證PVDF和NMP與P表面自然形成的氧化物層碳化的反應,將在室溫下氧化5天的黑磷(BP-5)與新鮮黑磷(BP)進行比較。BP表現出均勻和松散的分布,而BP-5表現出嚴重的團聚現象(圖1c,d)。將這兩個樣品分別加入含有3%PVDF的NMP和NMP中,攪拌過夜,然后用環己烷洗滌3次,以排除殘余PVDF和NMP對樣品的影響。
之后,將所得產物過濾并在真空中干燥以進行STEM線掃描。對于BP-5在NMP-3%PVDF溶液中的情況(圖1e,f),分別有來自NMP的C和N元素、來自PVDF的F元素以及來自氧化磷的P和O元素的信號。C元素的分布比P和O元素的分布更廣,表明NMP和PVDF在黑磷表面會發生碳化反應。
因此,漿料制備過程中碳化層形成的示意圖如圖1g所示。碳化反應可以增強磷與粘結劑的相互作用,有利于降低界面阻力,提高循環穩定性。
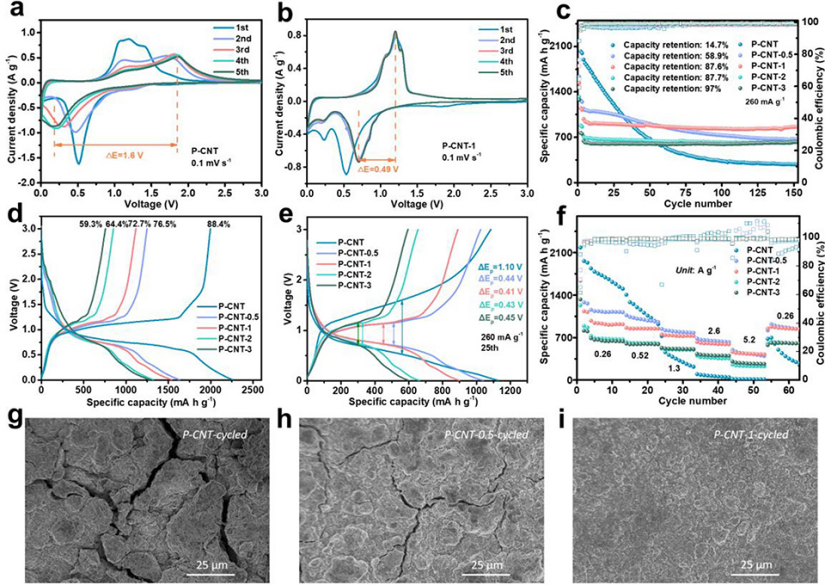
圖 2、(a)P-CNT和(b)P-CNT-1負極的CV曲線,掃速為0.1 mV s–1。(c)長循環穩定性。(d)50 mA g–1下的初始充放電曲線。在260 mA g–1下,(g)P-CNT、(h)P-CNT-0.5和(i)P-CNT-1電極25次循環后的SEM圖像。
圖2a顯示,在第一次陰極掃描中,原始P-CNT負極在~1.16 V處顯示了一個寬的不可逆峰,這是由于電解質的不可逆分解形成SEI。0.52 V的另一個還原峰對應于從P到LixP化合物的逐步鋰化過程(x=1–3)。在第一次陽極掃描中,1.19V處出現陽極峰,這源于LixP的逐步脫鋰過程。
在隨后的循環中,陰極峰逐漸移動到低電壓,而陽極掃描的峰位置逐漸移動到高電壓,峰面積減小,表現出遞增的極化和較差的循環穩定性。而P-CNT-1(圖2b)負極由于其小的過電位、隨后的CV曲線高度重疊,表現出高的可逆性。
圖2c顯示,原始P-CNT在第一次循環中表現出2025.1 mAh g–1的高初始比容量,但在150次循環后,容量逐漸下降至279 mAh g-1,保留率僅為14.7%。P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的初始放電比容量分別為1632.4、1536.6、1322.2和1278.1 mAh g–1。由于氧化層的產生導致活性磷的一定損失,初始容量隨著氧化期的增加而逐漸減小。
此外,初始庫侖效率(ICE)從原始電極的88.4%逐漸降低到P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的76.5%、72.7%、64.4%和59.3%(圖2d)。在260 mA g–1下循環150次后,P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的容量保持率分別增加到58.9%、87.6%、87.7%和97%。圖1e顯示,P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的充放電電壓極化(ΔEp)分別為0.44、0.41、043和0.45 V,明顯小于原始P-CNT(1.10 V),表明氧化樣品具有降低的內阻。
倍率性能如圖2f所示。盡管P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的初始放電容量顯著低于原始P-CNT,但當提高電流密度時,前者的容量衰減較小。因此,表層氧化物層的存在有利于提高倍率性能。
通過SEM表征了25次循環后P-CNT、P-CNT-0.5和P-CNT-1電極的表面形貌。由于體積變化較大,原始P-CNT電極中存在嚴重裂紋(圖2g)。對于P-CNT-0.5電極(圖2h),盡管電極表面的完整性得到了改善,但由于磷顆粒的表面氧化不足,仍然可以觀察到一些微小的裂紋。
相反,P-CNT-1電極(圖2i)保持致密和光滑,沒有明顯斷裂。P-CNT、P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3的ICE隨著氧化時間的增加而逐漸減少(圖2d)。這種不可逆的容量損失主要來自SEI形成的消耗。
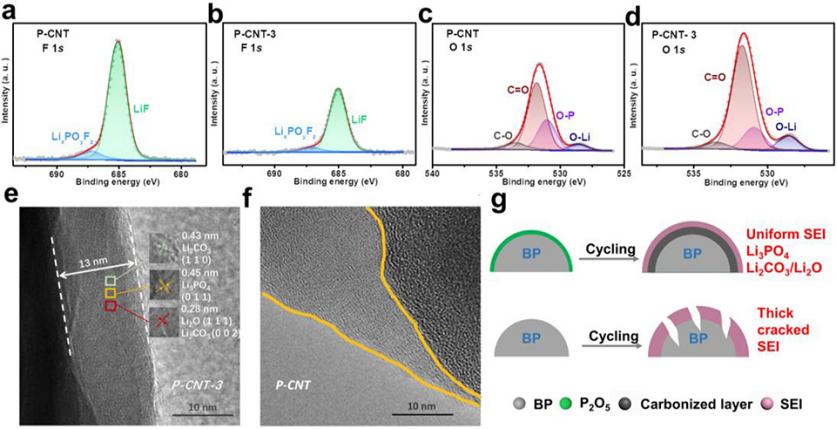
圖 3、150次循環后,P-CNT和P-CNT-3電極的高分辨率F 1s(a)、(b)和O 1s(c)、(d)XPS光譜。(e)P-CNT和(f)P-CNT-3電極的高分辨率TEM圖像。(g)SEI形成過程示意圖。
150次循環后,P-CNT和P-CNT-3的F 1s和O 1s XPS光譜如圖3a–d所示。F 1s光譜可以擬合為LiF(685.1eV)和LixPOyFz(687.1eV),它們是電解質分解的副產物。與P-CNT(圖3a)相比,P-CNT-3(圖3b)的F 1s光譜中LiF和LixPOyFz峰面積更低,表明氧化物層可以有效減少電解質與活性磷的副反應。O1s可以擬合成O–Li(528.6 eV)、O–P(531.0 eV)、C═O(531.8 eV)和C–O(533.5 eV)鍵。O 1s光譜中的O–Li信號主要對應于SEI中具有優異離子電導率的Li3PO4、Li2O和Li2CO3。P-CNT-3的O1s光譜中O–Li鍵的峰面積明顯大于原始P-CNT,表明P-CNT-3中產生了高含量的Li3PO4、Li2O和Li2CO3。
通過TEM表征了150次循環后P-CNT和P-CNT-3電極的SEI形態(圖3e,f)。P-CNT-3在循環后呈現出均勻的SEI層,約13nm,其主要包含Li3PO4、Li2CO3和Li2O。而原始P-CNT的SEI層呈現出不均勻的分布。圖3g顯示,富含Li3PO4和Li2CO3的SEI不僅有助于緩解體積膨脹,而且提高了反應動力學。
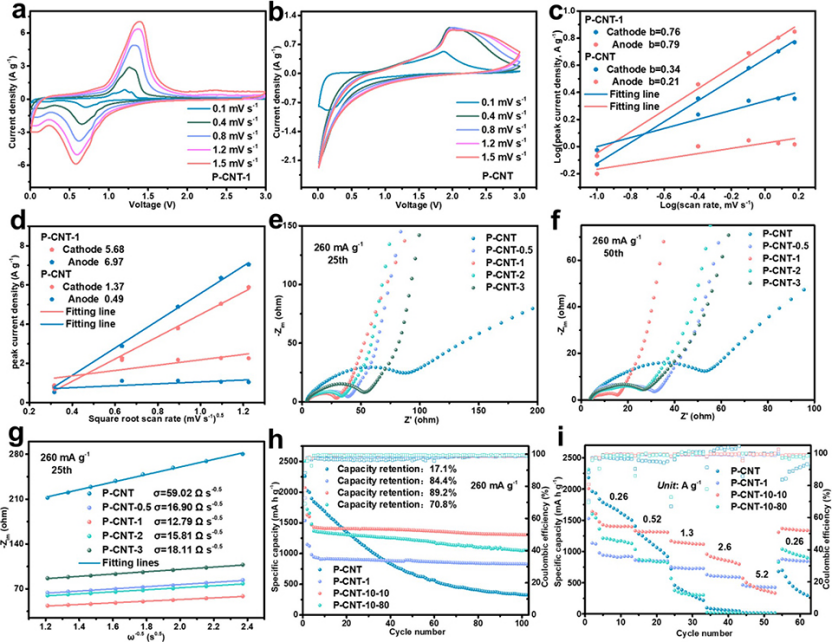
圖 4、在0.1至1.5 mV s–1不同掃速下,LIB的(a)P-CNT-1和(b)P-CNT的CV曲線。(c)陰極和陽極峰的Log(峰電流)–Log(掃速)曲線。(d)P-CNT-1和P-CNT負極陰極和陽極峰的Ip/ν1/2曲線。P-CNT, P-CNT-0.5, P-CNT-1, P-CNT-2和P-CNT-3電極在循環(e)25,(f)50圈之后的EIS譜,以及循環25圈后角頻率均方根倒數與低頻下?Zim的關系。P-CNT、P-CNT-1、P-CNT-10-10和P-CNT-10-80的(h)長循環穩定性和(i)倍率性能。
進行CV測試以研究不同掃速的動力學因素(圖4a,b)。方程1和2用于研究反應動力學過程。

其中,i表示峰電流(mA),ν表示掃速(mV s–1),a和b表示可變參數。
P-CNT-1負極的陰極(0.76)和陽極(0.79)峰b值明顯大于P-CNT負極(0.34和0.21)(圖4c),表明前者具有更好的動力學性能。此外,利用Randles–Sevcik方程(方程式3)研究擴散系數。
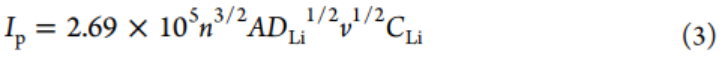
其中Ip、n、A、DLi、CLi和v表示峰電流、電荷轉移數、電極面積、Li離子擴散系數、Li離子濃度和掃速。P-CNT-1和P-CNT電極的DLi可以通過線性擬合Ip與v1/2來評估。P-CNT-1電極的陽極和陰極峰的Ip/v1/2值分別為6.97和5.68(圖4d),遠大于P-CNT的值(0.49和1.37),表明P-CNT-1電極在原始P-CNT上具有更快的Li離子擴散。
分別在260 mA g–1下進行1次、25次、50次和100次循環后,獲得了P-CNT、P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3電極的EIS光譜。如圖4e、f所示,氧化樣品(P-CNT-0.5、P-CNT-1、P-CNT-2和P-CNT-3)在1、25、50和100次循環后的電荷轉移電阻明顯小于P-CNT,表明氧化物層可以有效地降低界面電荷轉移電阻。
方程4和5用于計算Li+擴散系數。
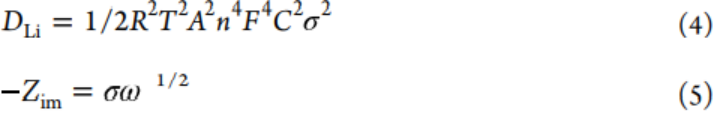
其中,氣體常數、絕對溫度、電極面積、法拉第常數和頻率分別用R、T、A、F和ω表示。電池阻抗的虛部和低頻區域的Warburg因子分別用?Zim和σ表示。因此,利用由?Zim與ω-1/2擬合得到的斜率Warburg系數σ來評價離子電導率。所有氧化樣品的σ值均低于原始P-CNT的σ值,無論其是否經過1、25、50和100次循環(圖4g),并且P-CNT-1在四個氧化負極中具有最小的σ,表明其具有優異的離子傳導性和中等厚度的SEI。
為了探索氧化層對P顆粒的保護作用,將P-CNT電極置于10%低濕度環境中1天,以在電極表面形成氧化物層。之后,將氧化電極分別置于10%低濕度(定義為P-CNT-10-10)或80%高濕度(定義P-CNT-10-80)環境中再放置1天。P-CNT-10-10的電極提供1306 mAh g–1的比容量,并在100次循環后,保持89.2%的容量保持率(圖4h)。
P-CNT-10-80的電極表現出較差的循環性能,因為當其置于高濕度條件下時,氧化層由于吸附的水而被破壞。P-CNT-10-10的倍率性能高于P-CNT和P-CNT-10-80。P-CNT-10-10-10的容量和循環性能高于P-NT-1,表明電極氧化法明顯優于粉末氧化法。
05
總結與展望
為了解決磷負極體積膨脹大、SEI層不穩定、空氣和水穩定性差等問題,本工作提出了在磷表面原位包覆氧化層的策略。氧化層不僅可以起到保護的作用,延長磷負極在空氣中的儲存時間,還可以碳化NMP和PVDF,形成碳化層,加強磷顆粒和粘結劑之間的界面相互作用。生成的氧化層可以進一步誘導形成穩定、均勻和高Li+電導率的SEI。
氧化的P-CNT保持1306 mAh g–1的高比容量,100個循環后的容量保持率為87%,遠高于原始P-CNT(17.1%)。這種原位氧化法簡單,沒有額外的材料或加工成本,有望實現工業生產。
審核編輯:劉清
-
鋰離子電池
+關注
關注
85文章
3215瀏覽量
77565 -
SNMP
+關注
關注
0文章
83瀏覽量
29721 -
固體電解質
+關注
關注
0文章
46瀏覽量
8381
原文標題:天大孫潔Nano Lett.:磷表面氧化提高儲鋰性能
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
贛鋒鋰業布局長時儲能市場,瞄準大容量電池
堿錳電池的銅針表面怎么處理
億緯鋰能閃耀SNEC 2024,展現全系列儲能解決方案
【古瑞瓦特光伏逆變器品牌】一文讀懂PCS儲能變流器

巖土工程監測中振弦采集儀的布設方案及實施步驟簡析

簡析智慧燈桿一鍵告警功能的實用場景

壓縮空氣儲能和二氧化碳儲能的區別
簡析電氣火災的原因及其對策
【鴻蒙】OpenHarmony 4.0藍牙代碼結構簡析
簡析電動汽車充電樁檢測技術應用及分析
如何提高鋰電儲能系統地位?
儲能的三大應用場景簡析






 工商網監
工商網監
評論