") PtNi納米合金可與Ni單原子發(fā)生原子置換
PtNi納米合金可與Ni單原子發(fā)生原子置換
研究背景
多相金屬催化劑在化工生產、環(huán)境保護和可持續(xù)能源生產中發(fā)揮著重要作用。為了實現催化活性金屬的有效利用,金屬物種通常被設計為高度分散的形式,如金屬納米顆粒、納米團簇和單原子形式。探索催化活性金屬物種在載體上的轉化途徑對于制備高效催化劑、研究催化劑如何失活以及廢舊催化劑的再生至關重要。目前,燒結和再分散是多相催化劑中金屬活性組分的兩種主要轉化方式,分別代表金屬活性物質在載體上的自下而上和自上而下的轉換。
成果簡介
鑒于此,清華大學李亞棟院士與北京師范大學李治教授(共同通訊作者)等發(fā)現,PtNi納米合金中的Ni與ZIF-8衍生氮摻雜碳(Zn1-CN)載體上的Zn在高溫下發(fā)生了金屬原子交換,生成PtZn納米晶和Ni單原子(Ni1-CN)。(PtZn)n/Ni1-CN多位點催化劑在CO2還原反應(CO2RR)中表現出較低的CO2質子化和CO脫附能壘,大大提高了CO2RR的催化性能。
研究亮點
1、發(fā)現了一種新型金屬催化劑,通過金屬原子在單原子和納米合金之間的交換作用,形成了一套新的納米合金和單原子;
2、PtNi納米合金可與Ni單原子發(fā)生原子置換,這種獨特的原子置換轉化也適用于其他金屬合金,如PtPd等。
圖文介紹
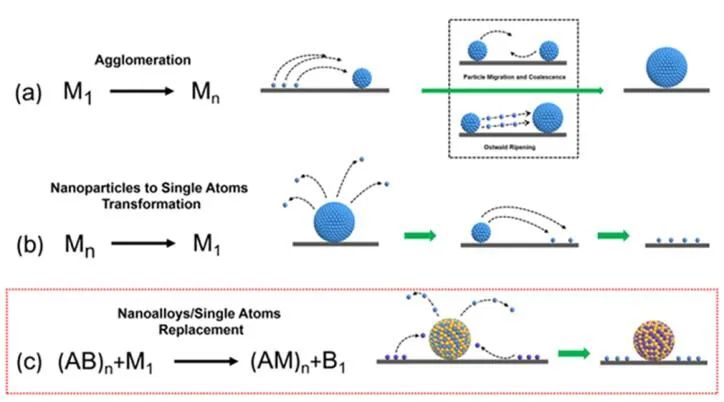
圖1單原子和納米粒子之間的轉化途徑。(a)單個原子凝聚成納米粒子;(b)納米顆粒再分散(單原子化)形成單個原子;(c)單原子和納米合金之間的原子替換。
顆粒遷移/聚結和奧斯特瓦爾德熟化是金屬燒結的兩種主要機理。納米粒子遷移/聚結表示納米顆粒的移動和結合會形成更大的聚集體,奧斯特瓦爾德熟化表示原子級物種會從較小的納米顆粒遷移到較大的納米顆粒(圖1a)。與燒結相反,再分散減小了金屬活性組分的尺寸,增加了多相催化劑表面原子的比例。
目前,金屬再分散可以實現金屬納米顆粒或大塊金屬到金屬單原子的轉化(單原子化)(圖1b)。再分散/單原子化通常會顯著提高催化性能,這也是實現廢舊催化劑再生的重要途徑。作者發(fā)現了一種全新的多相金屬催化劑轉化模式,即單原子與納米合金之間的原子置換,即通過單原子與納米合金之間的原子置換,形成單原子與納米合金的新組合(圖1c)。
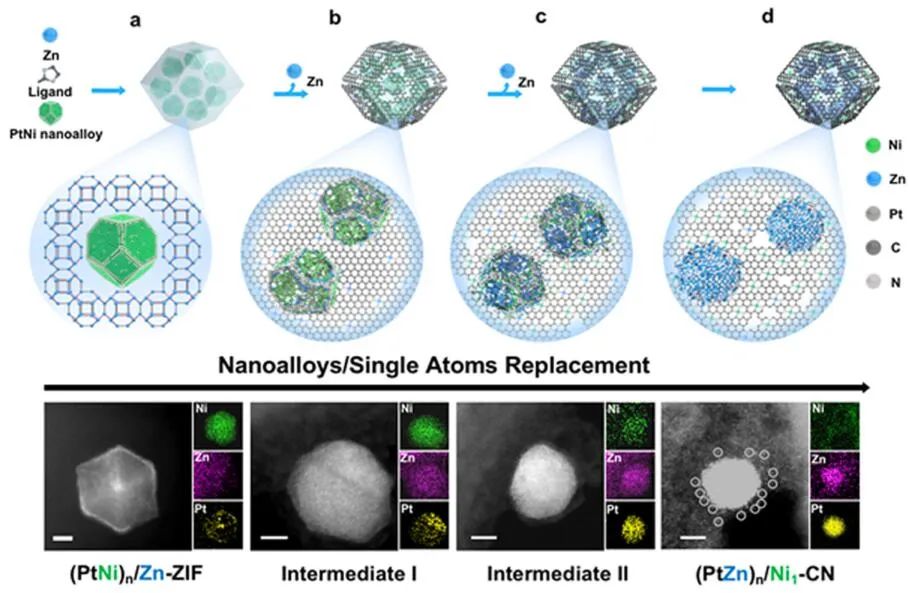
圖2納米復合材料的原子置換及結構表征。(a-d)(PtNi)n/Zn-ZIF(a)、中間體I(b)、中間體II(c)、(PtZn)n/Ni1-CN(d)的AC-STEM圖像及其對應的EDS元素映射圖。
在含有分散的PtNi納米合金的Zn(NO3)2溶液中加入2-甲基咪唑溶液,制備了PtNi納米合金/ZIF-8納米復合材料。ZIF-8納米晶體在PtNi納米合金周圍生長,生成(PtNi)n/Zn-ZIF納米結構。EDS元素映射和元素線掃描顯示,Zn作為ZIF-8的組成元素均勻分布在納米復合材料中,而Pt和Ni元素僅存在于PtNi納米合金上(圖2a)。
當加熱到600°C時,原始PtNi多面體結構的邊角變得模糊,Zn開始從襯底滲透到納米合金中(圖2b)。在700~800℃時,這一趨勢更加明顯,Ni原子從金屬納米合金擴散到載體上(圖2c)。Ni和Zn在900℃完成原子置換。在原子置換過程中,Zn原子從襯底富集到納米合金顆粒,Ni原子從納米合金遷移到CN載體,Pt原子自始至終分布在納米合金顆粒上(圖2a?d)。
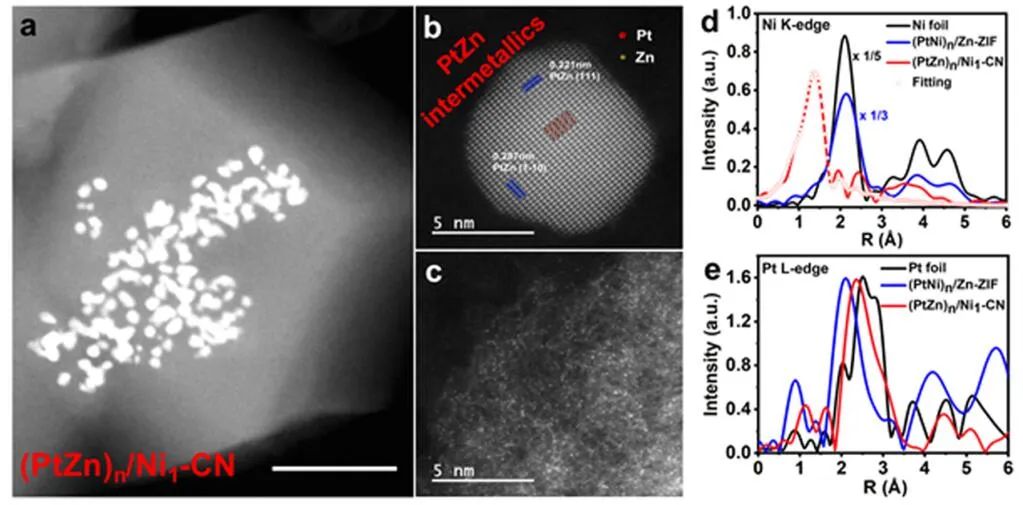
圖3 (PtZn)n/Ni1-CN的結構表征。(a)(PtZn)n/Ni1-CN的HAADF-STEM圖像;(b, c)單個PtZn納米晶體和(b)CN襯底上原子分散的金屬原子的AC-STEM圖像(c);(PtNi)n/Zn-ZIF和(PtZn)n/Ni1-CN中Ni K邊(d)和Pt L邊(e)的FT-EXAFS譜。
作者對900℃熱解后的產物(PtZn)n/Ni1-CN進行了表征。HAADF-STEM和TEM圖像顯示CN襯底發(fā)生收縮,納米合金內部尺寸減小(圖3a)。從像差校正掃描透射電鏡(AC-STEM)圖像分析,納米晶相具有清晰的晶格條紋,晶格間距分別為0.221和0.287 nm(圖3b),分別對應于金屬間化合物PtZn的(111)和(1-10)平面,表明Zn摻雜到納米合金相中,生成了PtZn金屬間化合物。
(PtNi)n/Zn-ZIF在Ni K邊的傅里葉變換EXAFS(FT-EXAFS)曲線顯示了一個約2.21 ?的峰,來自Ni?M(M=Ni或Pt)散射,而(PtZn)n/Ni1-CN樣品中原子分散的Ni只有一個約1.44 ?的峰,歸因于Ni?N/C散射,驗證了原子替換后原子分散的Ni原子的存在(圖3d)。在Pt L邊的FT-EXAFS曲線(圖3e)中,(PtNi)n/Zn-ZIF的最近配位峰出現在2.12 ?處。Zn原子取代Ni原子后,得到的(PtZn)n/Ni1-CN由于Pt-Zn鍵長比Pt-Ni更長,其R空間位于2.33 ?,而(PtNi)n/Zn-ZIF的R空間位于2.12 ?。
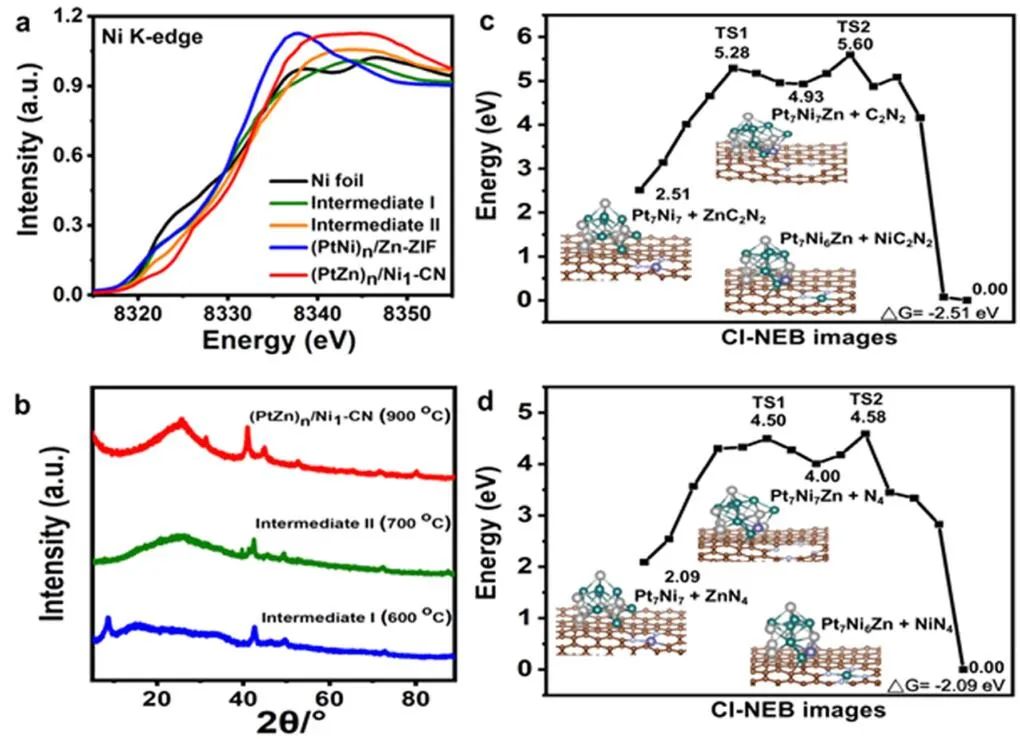
圖4 (a,b)不同熱解溫度下,樣品Ni k邊的XANES(a)和XRD譜圖(b);(c,d)采用CI-NEB方法計算路徑上的能量分布,以及不同配位環(huán)境下相應的初始態(tài)、最終態(tài)和過渡態(tài)構型。
X射線吸收近邊結構譜(XANES)表明,熱解過程中,(PtNi)n/Zn-ZIF中Ni的價態(tài)隨著原子置換的進行逐漸增加(圖4a),表明PtNi的解構和Ni1-CN的形成。熱解后,(PtNi)n/Zn-ZIF的XRD譜圖沒有顯示Ni團簇或納米顆粒的峰(圖4b)。 作者通過密度泛函理論(DFT)計算,研究原子替換的機理(圖4c,d)。從EXAFS(圖3d)可以看出,(PtZn)n/Ni1-CN中Ni單原子為四配位,因此分別建立了ZnCxN4?x和Pt7Ni7模型來模擬Zn單原子和PtNi納米合金。
從Pt7Ni7+ZnC2N2模型到Pt7Ni6Zn1+NiC2N2的交換能為?2.51 eV;從Pt7Ni7+ZnN4模型到Pt7Ni6Zn1+NiN4的交換能為?2.09 eV。原子交換過程中,經歷兩個階段:第一步,Zn從ZnCxN4?x轉移到PtNi合金中;第二步,Ni與CxN4?x結合。C2N2缺陷與Ni具有較大的結合能,在高溫下克服反應能壘時,原子取代產物應以NiC2N2為主,而不是Ni-N4。
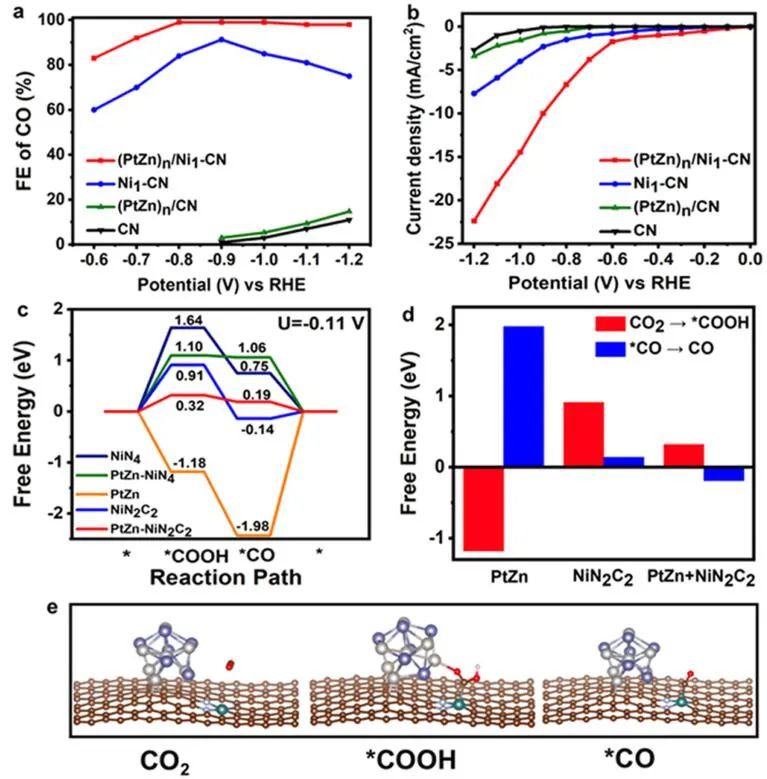
圖5 不同催化劑對CO2RR的催化性能和密度泛函理論計算。(PtZn)n/Ni1-CN、(PtZn)n/CN、Ni1-CN和CN催化劑上CO電還原反應的法拉第效率(a)和不同電位下的LSV曲線(b);(c)CO2RR過程中的自由能變化;(d)CO2RR中關鍵步驟的自由能比較;(e)(PtZn)n/Ni1-CN催化CO2RR過程中多位點協同模型的中間結構優(yōu)化。
當CO2電還原為CO時,合成的(PtZn)n/Ni1-CN與Ni1-CN、PtNi納米合金和(PtZn)n/CN相比表現出顯著增強的性能。(PtZn)n/CN和CN在電位范圍為-0.9至-1.2 V時,CO的法拉第效率(FE)小于25%。單原子催化劑Ni1-CN在?0.9 V下FE可達90%。但隨著工作電壓的增加,FE急劇下降。
相比之下,(PtZn)n/Ni1-CN多位點催化劑在寬的電化學窗口(-0.7到-1.2 V)內表現出更高的CO選擇性(>90%)。如線性掃描伏安(LSV)曲線所示(圖5b),CN在?1.0 V時提供的電流密度僅為0.5 mA cm-2。在此電壓下,(PtZn)n/Ni1-CN催化劑的電流密度為14.5 mA cm-2,遠高于Ni1-CN(4.0 mA cm-2)和(PtZn)n/CN(1.6 mA cm-2)樣品的催化活性之和,說明(PtZn)n/Ni1-CN中PtZn納米顆粒與Ni1-CN具有協同作用。
隨著電壓的增加,(PtZn)n/Ni1CN的電流密度急劇增加,遠遠超過Ni1-CN和(PtZn)n/CN。 如圖5c所示,四配位結構的Ni單原子位點形成*COOH中間體需要1.64 eV。當配位環(huán)境中的部分N替換為C時,*COOH在NiN4位點上的吸附能降低至0.91 eV,但在中等電位下仍難以達到。引入的PtZn納米晶體通過Pt-O相互作用對*COOH中間體起到了吸附和穩(wěn)定作用,PtZn-NiN2C2的質子化能量從0.91 eV顯著降低到0.32 eV。
但PtZn-NiN4的質子化能量仍然較高,這意味著*COOH中間體更容易在PtZn-NiN2C2位點上形成。另一方面,*CO的脫附對CO的生成也很重要。從熱力學角度看,盡管CO2在PtZn納米晶體上的直接質子化過程在能量上更有利,約為?1.18 eV,但最后一步對*CO的脫附而言非常困難。因此,與單個PtZn或Ni原子相比,多位點PtZn-NiN2C2可以同時促進質子化和*CO脫附(圖5d)。
總結與展望
本工作發(fā)現了單原子與納米合金之間發(fā)生原子置換的新現象,豐富了納米粒子與單原子之間的轉化策略。原子置換也為合成多活性位點催化劑建立了新的策略。DFT計算揭示了原子置換機理,并證明了制備的多活性位點催化劑具有協同催化作用。
審核編輯:劉清
-
DFT
+關注
關注
2文章
224瀏覽量
22681 -
傅里葉變換
+關注
關注
6文章
438瀏覽量
42566 -
XRD
+關注
關注
0文章
131瀏覽量
9061 -
EDS
+關注
關注
0文章
94瀏覽量
11515
原文標題:JACS:利用PtNi納米合金與Zn-ZIF-8之間的原子置換,制備多位點CO2還原電催化劑
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發(fā)布評論請先 登錄
相關推薦
使用Phase Lab2024A計算間隙原子擴散的非平衡凝固
微型原子鐘專用795nm VCSEL
正點原子和野火開發(fā)板哪個好
第二屆開放原子大賽火熱開啟
原子層鍍膜在功率器件行業(yè)的應用
磷酸二氫鋰的相對原子質量是多少
全球首款原子級精度的量子傳感器研發(fā)成功
通過 ORCA-Quest 成像單原子陣列以實現中性原子量子計算
原子力顯微鏡AFM測試與案例分享
原子結構示意圖次外層電子不超過多少
原子陣列實現容錯通用量子計算的前景和挑戰(zhàn)
開放原子云社區(qū)正式成立

原子級量子芯片如何制造的?
鴻智谷亮相2023開放原子開發(fā)者大會






 工商網監(jiān)
工商網監(jiān)
評論