 付超鵬Angew:聚合物基p型正極實現超長循環鋁電池
付超鵬Angew:聚合物基p型正極實現超長循環鋁電池
研究背景
鋁離子電池具有儲量豐富、成本低、安全性好、理論容量高等優點,是一種很有前景的大規模儲能技術。但是,具有無機正極的鋁離子電池在循環過程中仍存在動力學緩慢、結構坍塌等問題。
成果簡介
近日,上海交通大學付超鵬副教授在Angew上發表了題為“Phenoxazine Polymer-based p-type Positive Electrode for Aluminum-ion Batteries with Ultra-long Cycle Life”的論文,提出了一種p型聚(乙烯基芐基-N-吩惡嗪)(PVBPX)正極用于AIBs。雙活性位點使PVBPX能夠在0.2 A g?1下提供133 mAh g?1的高容量。PVBPX的擴展π共軛結構、不溶性和無鍵重排的陰離子氧化還原化學實現了50000圈的超長循環壽命。實驗和理論結果均證明,AlCl4?離子能夠可逆地與PVBPX中的N位和O位配位/解離,從而實現電荷存儲。
研究亮點
(1)本文通過對AIBs的一步親核取代反應,設計并制備了p型正極材料聚乙烯基芐基-N-吩惡嗪(PVBPX)。
(2)PVBPX中的N和O雙活性位點實現了133 mAh g?1的高比容量。此外,接枝在聚合物上的雙活性位點可以顯著抑制聚合物在電解質中的溶解。
(3)PVBPX的不溶性,擴展的π共軛體系以及無鍵重排的陰離子氧化還原化學加快了反應動力學,并實現了超長的循環壽命。
圖文導讀
聚合物PVBPX的合成過程如圖1a所示,通過一步親核取代反應將吩惡嗪(PX)單體與線性聚(乙烯基芐基氯)接枝,以獲得PVBPX。圖1b的傅里葉變換紅外(FTIR)光譜證實,吩嗪單元成功接枝到聚合物支鏈上。吩惡嗪的特征吸收峰在1700至700cm?1范圍內。PX中3390 cm?1處的N-H基團特征峰在PVBPX中消失,表明親核取代后氨基上的氫原子被線性聚合物的芐基取代。結果表明,PX單體成功接枝到聚合物支鏈上。
PX單體顯示出高結晶度結構,然而,制備的PVBPX沒有顯示出明顯的衍射峰,并顯示出無定形結構(圖1c)。PX單體在約150°C下分解和升華,并在270°C下失去所有重量。然而,PVBPX在280°C的更高溫度下分解,且PVBPX聚合物在270°C時的重量損失小于2.5%,顯示出優異的熱穩定性(圖1d)。
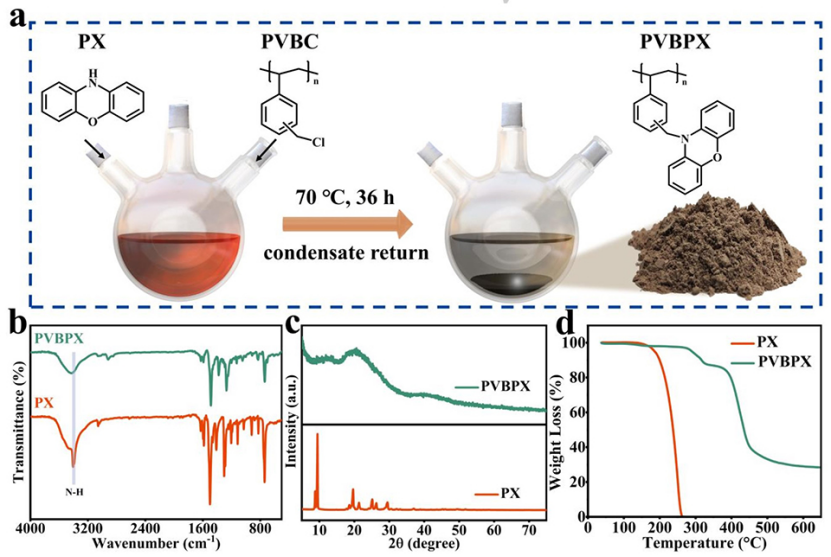
圖 1、PVBPX聚合物的(a)合成示意圖。(b)FT-IR光譜。(c)XRD圖譜。(d)PVBPX和PX在N2氣氛中的TG曲線。
以AlCl3/[EMIm]Cl離子液體電解質,PVBPX正極與Al負極組裝了AIBS,如圖2a所示。PVBPX正極的循環伏安法(CV)曲線在1.07/1.0V、1.81/1.72V附近出現兩對氧化還原峰(圖2b),表明PVBPX中活性位點發生了可逆的氧化還原過程。PX正極的CV曲線峰電位與PVBPX峰電位相似,說明PVBPX聚合物的氧化還原過程發生在吩惡嗪單元內。兩對氧化還原峰(O1/R1和O2/R2)可能分別歸因于-N和-O-活性位點上的氧化還原反應。帶有PX和PVBPX的AIBs恒流充放電曲線在1.0V和1.7V顯示出兩個不同的放電電壓平臺(圖2c)。
在0.2 Ag?1下,PVPBX電極放電容量為133 mAh g?1,首圈庫侖效率僅為83%,這與固體電解質界面的形成有關。圖2d顯示,PVBPX正極表現出優異的倍率性能。此外,即使在高電流密度下,PVBPX的充放電電壓平臺也保持良好(圖2e)。在200 mA g?1下,PVBPX正極循環100次后沒有容量衰減,而PX正極在100次循環之后,僅保持85%的容量(圖2f)。圖2g顯示,PVBPX正極在50000次循環后保持88 mAh g?1的比容量,每個循環沒有明顯的容量衰減,在50000次后庫侖效率保持99.8%。圖2h顯示,在循環50000次后,FTIR光譜中仍然檢測到C-N(1375和1265 cm?1)和C-O(1127 cm?1)的吸收峰,表明PVBPX的分子結構在長循環后保持完整。
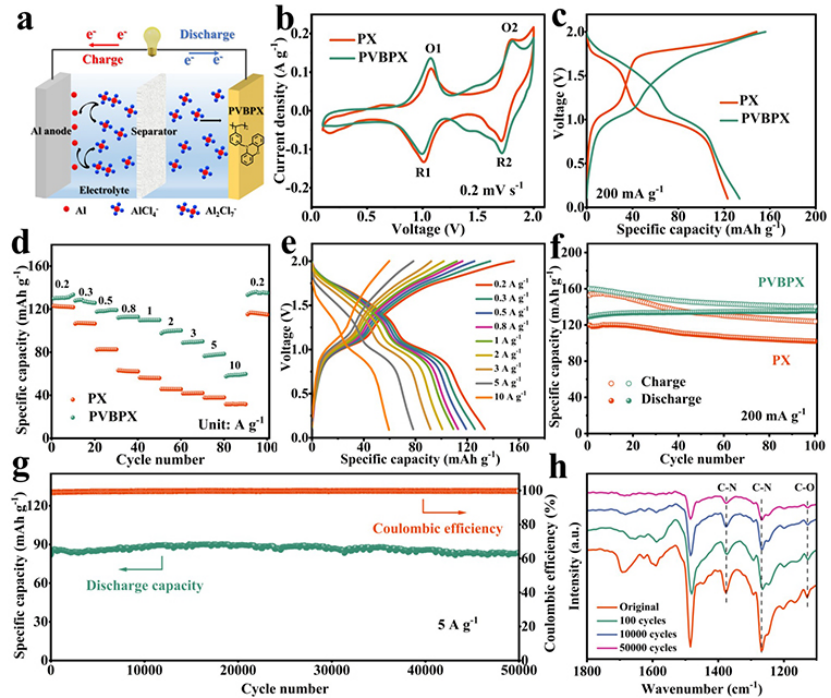
圖 2、(a)帶有PVBPX正極的AIBs示意圖。(b)具有PVBPX和PX的AIBs在0.2 mV s?1掃速下的CV曲線。(c)0.2 A g?1下的充放電曲線。(d)倍率性能和(e)各種電流密度下的充放電曲線。(f)PVBPX和PX在0.2Ag?1下的循環性能。(g)PVBPX和PX在5 A g?1下的長循環穩定性。(h)不同循環圈數下PVBPX電極的FTIR光譜。
圖3a中不同掃速下的CV曲線顯示,峰電流隨著掃速的增加而增加,峰值電流(i)和掃速(v)之間的關系可以寫為:
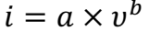
其中a和b為可調參數。b值可由log(i)與log(v)圖的斜率確定。如果b值為0.5,則表明充放電過程由離子擴散控制。如果b值為1.0,則電化學行為受表面電容支配。根據圖3b中的log(i)對log(v)圖,與四個峰O1、O2、R1和R2對應的b值分別為0.849、0.765、0.895和0.904。結果表明,PVBPX的電化學過程同時受到離子擴散和電容的影響。此外,表面電容貢獻和離子擴散貢獻的比率可以通過以下方程進一步計算。
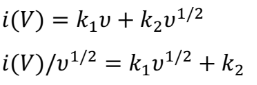
其中k1v和k2v1/2分別對應于表面電容和離子擴散貢獻。圖3c顯示,隨著掃速從0.2增加到5 mV s?1,PVBPX的表面電容貢獻從63.7%增加到89.1%,高于相應掃速下的PX電極(圖3d)。計算表明,PVBPX正極的充放電過程主要由表面贗電容貢獻,這促進了快速的電荷存儲,實現了出色的倍率性能。
圖3e的電化學阻抗譜(EIS)顯示,與PX電極相比,PVBPX電極在高頻區域顯示出更小的直徑,在低頻區域顯示出更陡的斜率,表明PVBPX電極具有小的電荷轉移電阻(Rct)和低的離子擴散電阻。PX和PVBPX電極的Rct在20次循環后都顯著降低,較小的Rct反映了高的電導率和快速的反應動力學。
圖3f顯示,PVBPX正極不僅提供了最佳的循環穩定性和倍率性能,還表現出具有競爭力的能量密度和放電電壓。因此,PVBPX是一種很有前途的可充電AIBs正極材料。
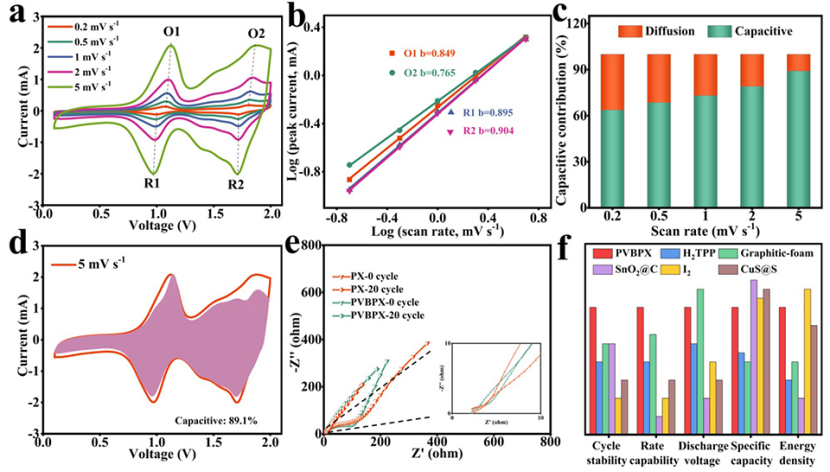
圖 3、(a)在0.2至5 mV s?1不同掃速下的CV曲線。(b)氧化還原峰處log(v)和log(i)的線性關系。(c)不同掃速下的電容貢獻。(d)5 mV s?1下的贗電容貢獻。(e)循環之前和20個循環之后的奈奎斯特圖。(f)AIBs中不同正極材料的關鍵電化學參數比較。
原位XPS光譜顯示,N1s光譜在399.4 eV處顯示出單峰(圖4b)。充電至1.4V后,峰向更高的結合能移動,在N1s光譜中401.4eV處出現新峰,表明C-N-C部分氧化為帶正電的C-N+-C。氧化的N+部分占總N原子的71.4%。充電至2.0V后,C-N+-C的分數達到72.5%。與充電至1.4V相比,氧化C-N+-C的分數沒有顯著差異,表明作為活性位點的氮原子在充電過程中被氧化,反應主要發生在1.4 V以下。
在O 1s光譜中可以觀察到類似的現象(圖4c),其中對應于氧化態的新峰出現在充電態的高結合能處。在充電狀態下,電荷離域導致N和O核的電子密度降低,這導致相應的XPS光譜向更高的結合能移動。與N 1s光譜不同,充電至1.4 V時,氧化峰(C-O+-C)的強度在O 1s光譜處較弱。然而,充電至2.0 V時,C-O+-C的峰明顯增強,表明O位的氧化電位高于N位。在隨后的放電過程中,氧化的C-N+-C和C-O+-C峰可逆地減弱,表明N和O活性位點表現出高的氧化還原可逆性。結合N 1s和O 1s光譜的分析,在~1.0 V的電壓平臺是N位的氧化還原反應,而在~1.7 V的電壓平臺則是O位的氧化反應。此外,Al 2p光譜的峰強隨著充電而逐漸增加,并在隨后的放電過程中可逆地減弱(圖4d)。雖然在Cl2p XPS光譜中觀察到類似的趨勢,但充放電過程分別伴隨著Cl信號的增強和減弱(圖4e)。這一結果表明,在充放電過程中,Al和Cl都參與了配位/解離反應。
圖4f的原位EPR顯示,在充電狀態下觀察到的g值為2.005,表明在充電過程中陽離子自由基的形成。同時,該結果證實陽離子PVBPX與帶負電的載體配合,進行陰離子氧化還原反應。在隨后的放電過程中,陽離子自由基的信號可逆地消失,進一步證實PVBPX的氧化還原過程是高度可逆的。
圖4g顯示,PVBPX正極的電荷存儲經歷兩步可逆過程。在充電過程中,N位首先被氧化成帶正電的狀態,同時與AlCl4-載流子結合。隨后,O位點被氧化形成自由基陽離子,該自由基陽離子也與AlCl4-發生配位反應。在放電過程中,陰離子的脫配位反應同樣是伴隨著氧化態逐漸還原的兩步反應過程。
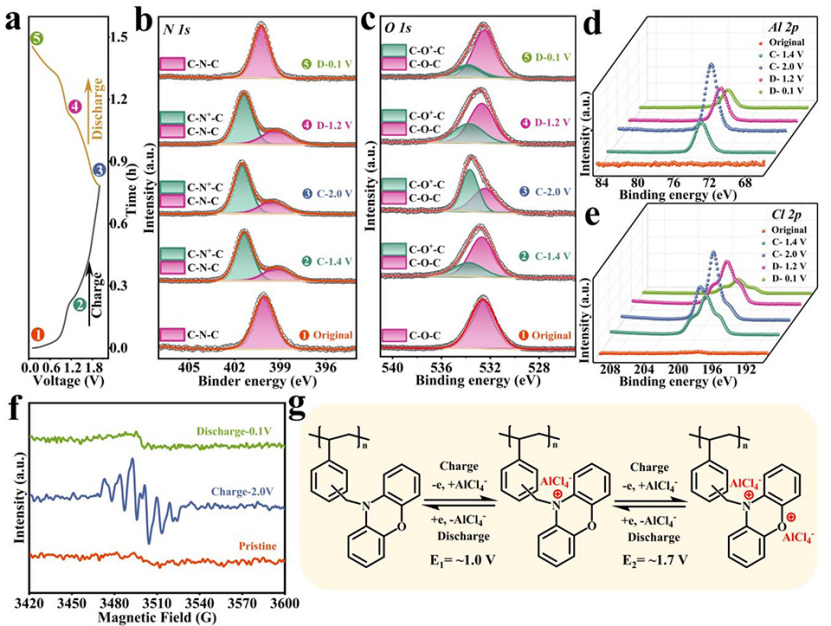
圖 4、(a)200 mA g?1下PVBPX正極的充放電曲線。(b)PVBPX正極在不同電壓下的N 1s、(c)O 1s、(d)Al 2p和(e)Cl 2p XPS光譜。(f)PVBPX電極的原位EPR譜。(g)PVBPX的電化學氧化還原機理。
中性PVBPX、單電子氧化產物(PVBPX+)和雙電子氧化產物(PVBPX2+)的分子靜電勢(ESP)分布圖,如圖5a所示。具有正靜電勢的藍色區域傾向于親核位點,并且容易參與親核反應。具有負靜電勢的紅色區域可以充當親電子位點,并傾向于進行親電子反應。在中性PVBPX的ESP圖中,雜原子兩側的兩個苯環區域表現出明顯的負電荷特征,具有強烈的給電子傾向。而氧化PVBPX+和PVPBX2+的ESP圖在整個分子中顯示出強烈的正電荷特性,特別是在中間雜環的N和O區域。ESP圖譜的計算結果從理論上證明,N和O是PVBPX分子中親核反應的活性位點,并與電解質中帶負電的載體配位。
此外,通過誘導電流密度(AICD)的各向異性研究了PVBPX及其氧化產物的芳香性變化。如圖5b所示,中性PVBPX分子的N-O雜環兩側的苯環表現出強烈的芳香特征,而中心N-O雜環表現出顯著的反芳香特征。在PVBPX分子失去一個電子后,N-O雜環從反芳香特性轉變為非芳香特性。失去另一個電子后,PVBPX2+的AICD圖呈現順時針循環,N-O雜環轉變為芳香性。這種反芳香族轉化遵循Huckel法則,在PVBPX的兩個氧化還原過程中只有π電子離域,這表明氧化中間體PVBPX+和PVBPX2+都是熱力學穩定的。
基于DFT計算了最低未占分子軌道(LUMO)和最高已占分子軌道的能級(HOMO)(圖5c)。中性PVBPX的前沿分子軌道居群及其氧化態表明,只有π-電子參與氧化還原反應過程,沒有觀察到鍵重排,證實了PVBPX在氧化還原反應期間的結構穩定性。此外,與中性PVBPX相比,PVBPX+和PVBPX2+的能隙明顯更低,這有助于充電期間PVBPX與陰離子的相互作用,促進電子轉移,并減少循環期間的極化。
此外,通過DFT優化了PVBPX分子及其與AlCl4?配位的配合物構型(圖5d)。在充電過程中,一個AlCl4?首先與PVBPX中的N原子配位,然后位于遠離乙基側鏈的一側。隨著進一步充電,第二個AlCl4?與O原子相互作用,然后位于雜環平面的另一側,使總能量最小化。放電過程是充電過程的可逆反應。
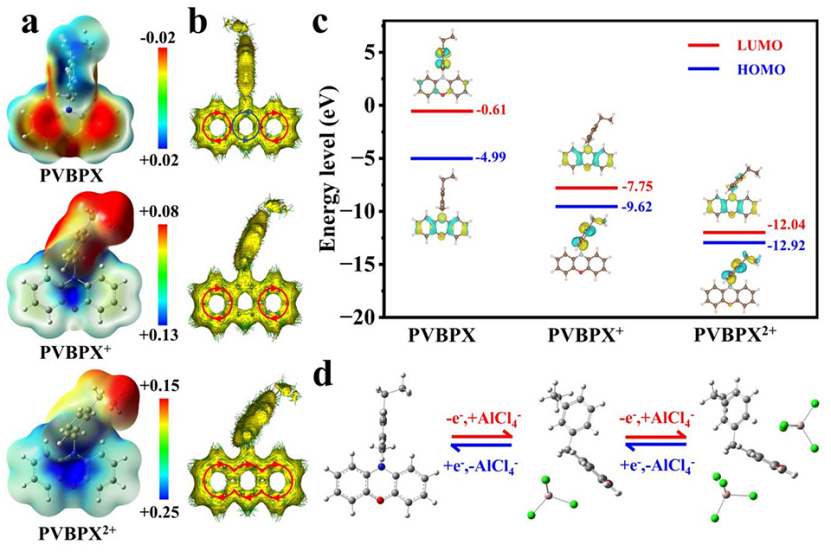
圖 5、PVBPX及其氧化物種(PVBPX+和PVBPX2+)的(a)分子ESP圖。(b)ACID圖。(c)LUMO-HOMO能級。(d)PVBPX電極的兩步氧化還原過程和分子幾何結構。
總結與展望
本工作成功開發了用于高性能AIBs的p型有機正極PVBPX。PVBPX中的雙活性位點有助于實現高比容量,設計的結構和獨特的配位反應保證了超長的循環壽命。各種表征技術和DFT計算表明,N和O是PVBPX中的雙氧化還原中心,在0.2 Ag?1下有133 mAh g?1的高比容量,擴展的π-共軛結構和無鍵重排的可逆陰離子氧化還原化學使得PVBPX能夠循環長達50000次。因此,p型有機聚合物是用于可充電AIBs的一種有前途的電極材料。
審核編輯 :李倩
-
正極
+關注
關注
0文章
52瀏覽量
10293 -
傅里葉變換
+關注
關注
6文章
437瀏覽量
42566 -
鋁離子電池
+關注
關注
1文章
11瀏覽量
4842
原文標題:付超鵬Angew:聚合物基p型正極實現超長循環鋁電池
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦





 工商網監
工商網監
評論