 闡明采用不同電解質的水系Zn/MnO2電池的反應機理
闡明采用不同電解質的水系Zn/MnO2電池的反應機理
研究背景
由于安全性和低成本,可逆水系Zn/MnO2電池(AZMOBs)成為一種有前途的電網級儲能替代方案。
近年來,由于需要充分了解其電荷存儲機制,以提高其效率和循環壽命,采用弱酸電解質的AZMOBs引起了人們的廣泛關注。
不同弱酸性電解質的AZMOBs中Mn的氧化還原機理目前仍存在爭議,主要有以下幾種:Zn2+/H+嵌入機制,Zn2+/H+嵌入誘導MnO2-ZnxMnOy/MnOOH轉換機制和Mnn+溶解?沉積機制。
最近,使用原位同步加速器X射線熒光譜(XFM)提供了MnO2溶解-沉積是ZnSO4電解質中主要的Mn氧化還原反應的直接實驗證據。然而,XFM方法無法提供結構信息,也無法提供固體正極或電解質內配位環境的演變信息。
成果簡介
鑒于此,石溪大學的Kenneth J. Takeuchi(通訊作者)等人利用Mn K邊XAS技術,對ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2水溶液的鋅電池的α-MnO2溶解-沉積氧化還原過程進行了實驗研究,此項技術能夠同時表征參與Mn氧化還原反應的液相和固相,最后闡明了采用不同電解質的水系Zn/MnO2電池的反應機理。
研究亮點
1、不同電解質(ZnSO4,Zn(CF3SO3)2和Zn(CH3COO)2)中,MnO2具有相似的錳配位環境,但固相和液相中Mnn+物種的定量分布存在差異;
2、拉曼光譜證明,在正極處,電荷作用下形成了結晶性差的含錳產物,并采用TEM對沉積物的形態和表面狀況進行了深入研究。
圖文介紹
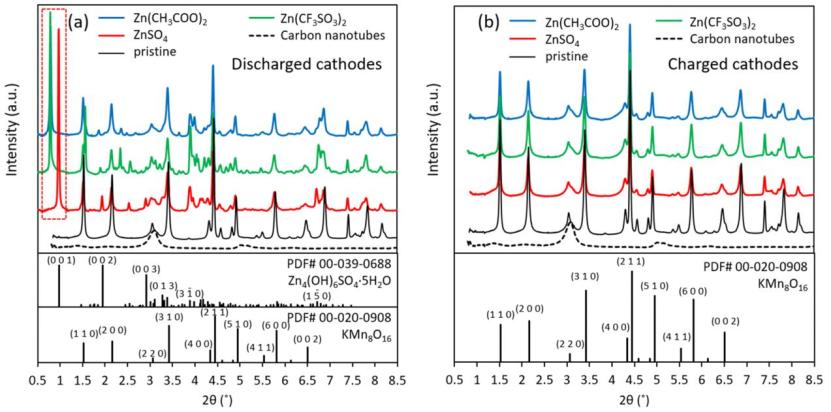
圖1 在ZnSO4、Zn(CF3SO3)2或Zn(CH3COO)2電解質中循環的(a)放電和(b)帶電正極與原始電極和碳納米管的同步X射線衍射圖。
采用基于同步輻射的XRD技術對不同電解質中循環至第一次放電和第一次充電的α-MnO2正極進行了表征(圖1)。
放電時,所有正極中都出現低于1°的強峰(圖1a紅色虛線框),表明形成了新相。以往文獻表明,ZnSO4,Zn(CF3SO3)2和Zn(CH3COO)2電解質中形成的新相分別為Zn4SO4(OH)6·xH2O(ZHS),Zn5(OH)8(CF3SO3)2·xH2O,(ZHT)和Zn5(OH)8(CH3COO)2·xH2O(ZHA)。
充電后正極的XRD圖譜中,ZHS、ZHA和ZHT峰消失,只留下原始材料的峰(圖1b),為未反應的α-MnO2。α-MnO2峰值強度較原始狀態電極的低,表明其質量分數降低。

圖2 在(a,d)ZnSO4電解質、(b,e)Zn(CF3SO3)2電解質和(c,f)Zn(CH3COO)2電解質中放電的α-MnO2正極的透射電鏡表征顯示了α-MnO2棒和沉積的板狀材料的圖像(a-c)和板狀材料的衍射圖(d-f)。
在電池初次放電(圖2)和初次充電(圖3)后,采用TEM觀測了α-MnO2正極。在首次放電時,三種電解質中的α-MnO2納米棒在表面和棒的末端附近都顯示出溶解現象(圖2a?c),在納米棒旁邊還觀察到板狀沉淀物質。ZnSO4電解液中放電產物的衍射模式與已知結構羥基硫酸鋅(ZHS)匹配性很好(圖2d)。

圖3 (a)ZnSO4,(b)Zn(CF3SO3)2和(c)Zn(CH3COO)2電解質中α-MnO2正極在首次充電循環后的TEM圖像顯示了充電過程中表面沉積的形成;(d-f)不同電解質下的嵌入功率譜。
ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2電解質中,第一個帶電電極的成像如圖3所示。三種電極類型的樣品均含有棒狀α-MnO2,電極表面鍍有電化學沉積物質,沉積的物質可能由含Zn/Mn相組成。
在圖3d-f中,嵌入功率譜中的紅色圈出的信號對應于Zn/Mn相。且過濾HRTEM圖像表明,表面沉積物都優先排列在α-MnO2棒上。
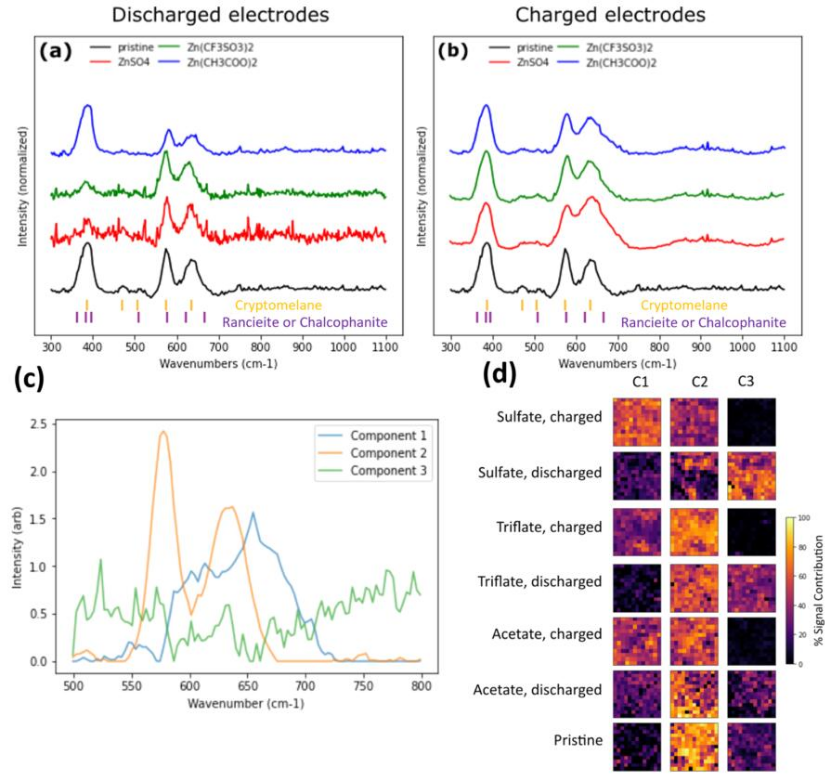
圖4 (a)放電電極和(b)帶電電極的拉曼光譜;(c)未混合共聚焦拉曼數據集的NMF分量。對提取的信號組分進行識別,其中組分1為層狀相,組分2為錳鉀礦,組分3為殘留背景;(d)非原位電極表面已識別組分的空間分布圖。
從原始電極上收集的拉曼光譜顯示,僅有與α-MnO2相關的峰,出現在387、471、494、507、577和635 cm?1。
在ZnSO4、Zn(CH3COO)2和Zn(CF3SO3)2電解質中放電電極的拉曼光譜與原始電極和α-MnO2材料相似(圖4a),表明放電電極中存在未反應的殘余α-MnO2。
充電后,在667 cm?1附近出現了一個新峰,表明形成了層狀Zn?Mn?O相(圖4b)。
對拉曼數據集進行NMF分量,其中組分1為層狀相,類似于層狀鈣硬錳石(Ca,Mn2+)0.2(Mn4+,Mn3+)O2·0.6H2O或黑鋅錳礦ZnMn34+O7·3H2O。
通過比較不同電解質和充放電條件下層狀相(成分1)的NMF歸一化信號強度,可以觀察到層狀相僅在充電電極中存在,這表明在充電過程中形成層狀相,在放電過程中消失(圖4d)。
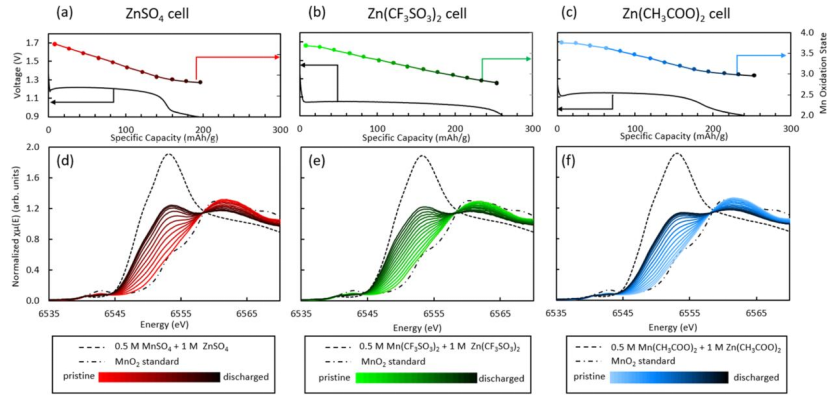
圖5 使用LCF計算的(a)ZnSO4,(b)Zn(CF3SO3)2,(c)Zn(CH3COO)2的操作單元放電期間的平均Mn氧化態的電壓分布;(d)ZnSO4,(e)Zn(CF3SO3)2和(f)Zn(CH3COO)2電池的原位XANES演化。
對每次XANES掃描進行線性組合擬合(LCF),利用KMn8O16(s)值和相應的溶液Mn2+標準,獲得電池的平均Mn氧化態(OS)。
在放電期間,XANES的X射線吸收邊緣轉移到較低的能量處(圖5d?f),這種變化表明平均Mn氧化態的降低,與由LCF確定的Mn平均氧化態變化一致(圖5a?c)。
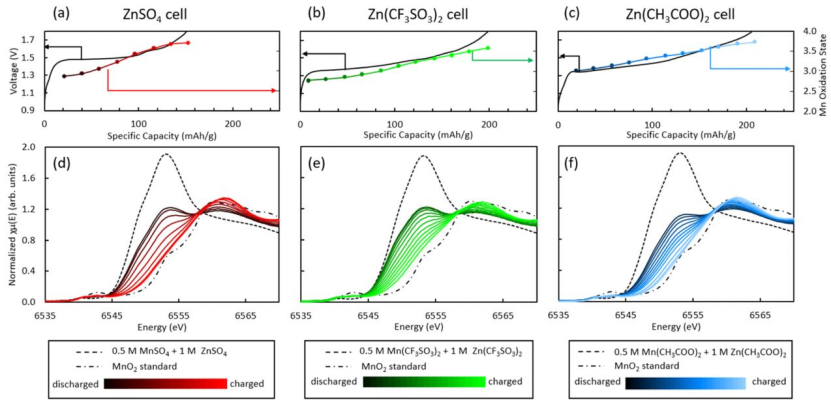
圖6 使用LCF計算的(a)ZnSO4,(b)Zn(CF3SO3)2,(c)Zn(CH3COO)2的操作單元充電期間的平均Mn氧化態的電壓分布。充電時,(d)ZnSO4,(e)Zn(CF3SO3)2和(f) Zn(CH3COO)2電池的原位XANES演化。
ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2電池在充滿電后對應的平均Mn氧化態值分別略低于原始材料值3.73、3.69和3.75。
這種氧化態變化歸因于充電產物與原始材料之間的結構和化學差異。如拉曼光譜表明,充電產物是層狀的鋅錳氧化物,這種材料通常由MnOx層組成,層間插入Zn2+離子。
與含有一價K+的原始α-MnO2相比,由于Zn2+的存在,使得放電產物具有較低的氧化態。
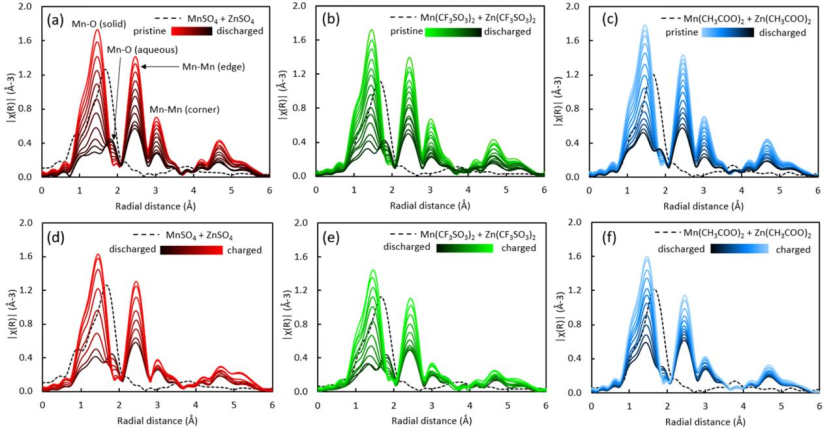
圖7 (a)ZnSO4,(b)Zn(CF3SO3)2和(c)Zn(CH3COO)2電池放電期間,原位EXAFS在r空間的演化;(d)ZnSO4,(e)Zn(CF3SO3)2和(f)Zn(CH3COO)2電池在充電過程中相應的EXAFS演化。
將原位EXAFS光譜傅里葉變換到徑向空間(r-空間),在r-空間中可以區分配位環境的變化。圖7顯示了在r-空間中初始放電和后續充電期間三個電池的原位EXAFS演化。
三種電池的r-空間EXAFS光譜具有相似的主峰。如圖7a所示,在~1.5 ?處的第一個主峰對應固體MnO2結構內的第一層Mn?O散射路徑,在~2.5 ?處的第二個主峰對應第二層Mn?Mn散射路徑,在~3.0 ?處的第三個主峰對應第三層Mn?Mn散射路徑。
第二層Mn-Mn散射路徑代表了MnO2結構中兩個共享邊的MnO6八面體的相對位置,而第三層Mn-Mn散射路徑代表了α-MnO2結構中兩個共享角的MnO6八面體的相對位置。
三個電池EXAFS光譜在初始放電過程中表現出相似的形狀,而始放電過程中,r空間峰值強度逐漸降低,并存在~1.9 ?處新峰的增長。與EXAFS標準Mn2+水溶液的對比表明,該峰可能對應于溶劑化[Mn(H2O)6]2+離子的第一層Mn?O散射路徑。
在充電時,除了第三層Mn-Mn峰外,所有的Mn-Mn峰都恢復到原始強度,三個電池的水Mn-O峰都消失了。
充電的α-MnO2與原始的α-MnO2在EXAFS光譜上的差異表明,在充電時,形成了一個具有不同于原始α-MnO2的Mn中心局部結構的產物。
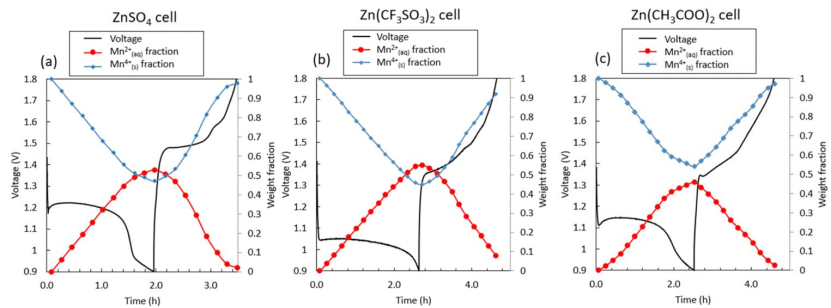
圖8 XANES-LCF結果顯示(a)ZnSO4,(b)Zn(CF3SO3)2和(c)Zn(CH3COO)2電池在第一個循環中的溶液中/固相中Mn的質量分數。
Mn的溶解-沉積過程包括溶解的溶劑化Mn2+離子和固體MnOx。使用標準Mn2+水溶液,對相應的電池進行原始掃描,以獲得電化學還原和氧化過程中單元內的水和固體Mn質量分數(圖8)。
在三種電池的初始放電過程中收集的XAS光譜表明,LCF擬合的固體/水Mn的重量分數呈現與電化學過程相關的Mn溶解現象(圖8)。
在初始放電結束時,約50%的Mn從正極溶解形成Mn2+。在充電過程中,雖然大多數溶解的Mn2+以固體形式重新沉積,但少量的Mn2+仍未被氧化。
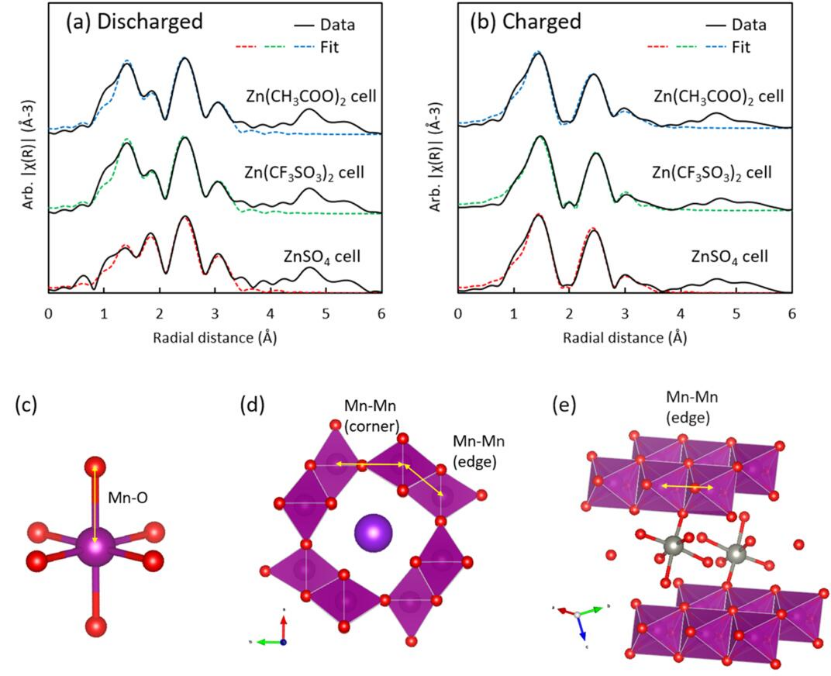
圖9 (a)完全放電和(b)充滿電的電池掃描的EXAFS擬合結果;(c?e)EXAFS擬合時用于執行FEFF計算的理論結構。
采用EXAFS擬合方法,利用理論FEFF計算結構對含錳產物進行解析。[Mn(H2O)6]2+結構可以用[MnO6]理論結構很好地表示(圖9c)。
初始放電時,原始α-MnO2溶解在ZnSO4、Zn(CF3SO3)2或Zn(CH3COO)2電解質中形成水合[Mn(H2O)6]2+離子,未溶解的α-MnO2沒有改變其局部結構。
對于充電電池的原位EXAFS光譜,放電時出現的Mn2+峰消失了,只在前三個殼層上留下三個主要峰。除了第三層Mn-Mn峰明顯減少,這些光譜與原始物相掃描光譜相似(圖7)。
這表明在充電正極中,Mn的角共享[MnO6]八面體的數量顯著減少。精細EXAFS擬合結果表明,在充電過程中,大部分Mn2+會轉化為固態ZMO,而未反應的KMO在電池的充電過程中都沒有發生結構變化。
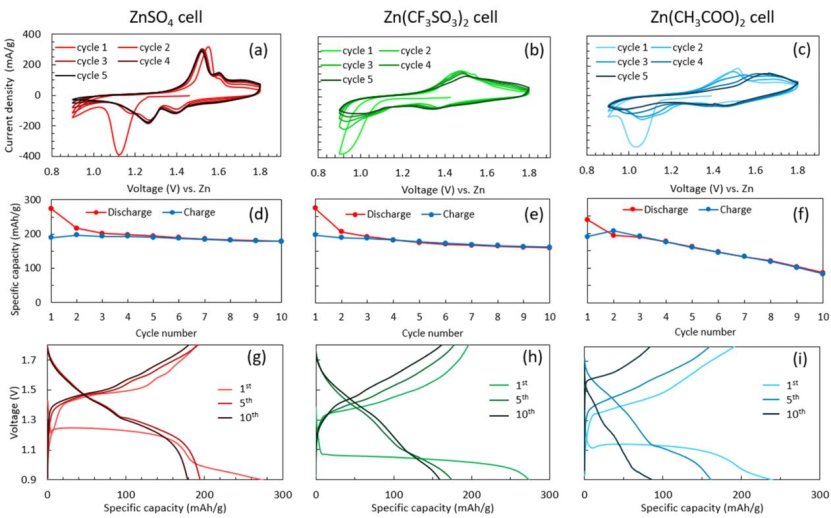
圖10 (a-c)5個周期的CV結果和(d-f)10個周期的容量保持曲線;(g,h)ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2電池在10個周期內的電壓分布演變。
三個電池的初始放電都以一個平坦的電壓平臺開始,但它們的開路電壓不同,ZnSO4電池顯示出最高的電壓。為了證明三種電解質體系之間的電化學差異,測試了這三種體系的CV(圖10a?c)和循環圖(圖10d?i)。放電平臺的差異很可能是由于陰離子效應造成的。
CF3SO3-陰離子具有更高的靜電電位,因此與SO42-陰離子(MPI=10.47 eV)相比,CF3SO3-陰離子具有更低的分子極性指數(MPI)(4.68 eV),這導致CF3SO3-陰離子具有更高的疏水性。因此,SO42?陰離子傾向于與H2O結合,在正極表面附近形成富水環境,而CF3SO3?和CH3COO?陰離子傾向于吸附在正極上,形成貧水環境。
這種貧水環境為錳溶解所需的質子化過程產生動力學障礙,降低了放電電壓平臺。相應地,在充電過程中,CF3SO3?或CH3COO?電池中體積較大的陰離子會減少Mn2+離子周圍的H2O水分子數量,減輕溶劑化效應,從而增強電荷和離子轉移,降低電荷電壓平臺。
對于Zn(CH3COO)2電池,在10個周期內觀察到明顯的容量衰退。這可能是由于CH3COO?和Zn2+離子之間的強結合,導致CH3COO?在充電時從ZHA中緩慢提取,因此限制了Mn2+插入到ZHA中,最終抑制了Mn的沉積。
總結與展望
本文采用Mn K邊原位XAS技術,對ZnSO4、Zn(CF3SO3)2和Zn(CH3COO)2水溶液鋅電池的α-MnO2溶解-沉積氧化還原過程進行了實驗研究。
EXAFS數據的分析采用了多相方法,分析了固態和溶解的過渡金屬成分。結果表明:三種弱酸性電解質的錳溶解-沉積過程具有相似的配位環境,但錳在固相和溶液中的分布不同。
原位XAS表征方法可以獨立研究固相或電解質中的反應。該方法為在復雜環境中對結晶性差的多相材料的表征提供了一種實用方法。
審核編輯:劉清
-
加速器
+關注
關注
2文章
795瀏覽量
37772 -
電解質
+關注
關注
6文章
805瀏覽量
20019 -
XRD
+關注
關注
0文章
131瀏覽量
9061
原文標題:JACS:闡明Zn/MnO2電池中固液錳環境
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
通過電荷分離型共價有機框架實現對鋰金屬電池固態電解質界面的精準調控
一種創新的超薄固體聚合物電解質
固態電池中復合鋰陽極上固體電解質界面的調控

無極電容器有電解質嗎,無極電容器電解質怎么測
電解質電極信號采集控制板

請問聚合物電解質是如何進行離子傳導的呢?
不同類型的電池的電解質都是什么?
開發一種生物兼容性水系Zn-MnO2電池正極—生物質碳集成策略

新型固體電解質材料可提高電池安全性和能量容量
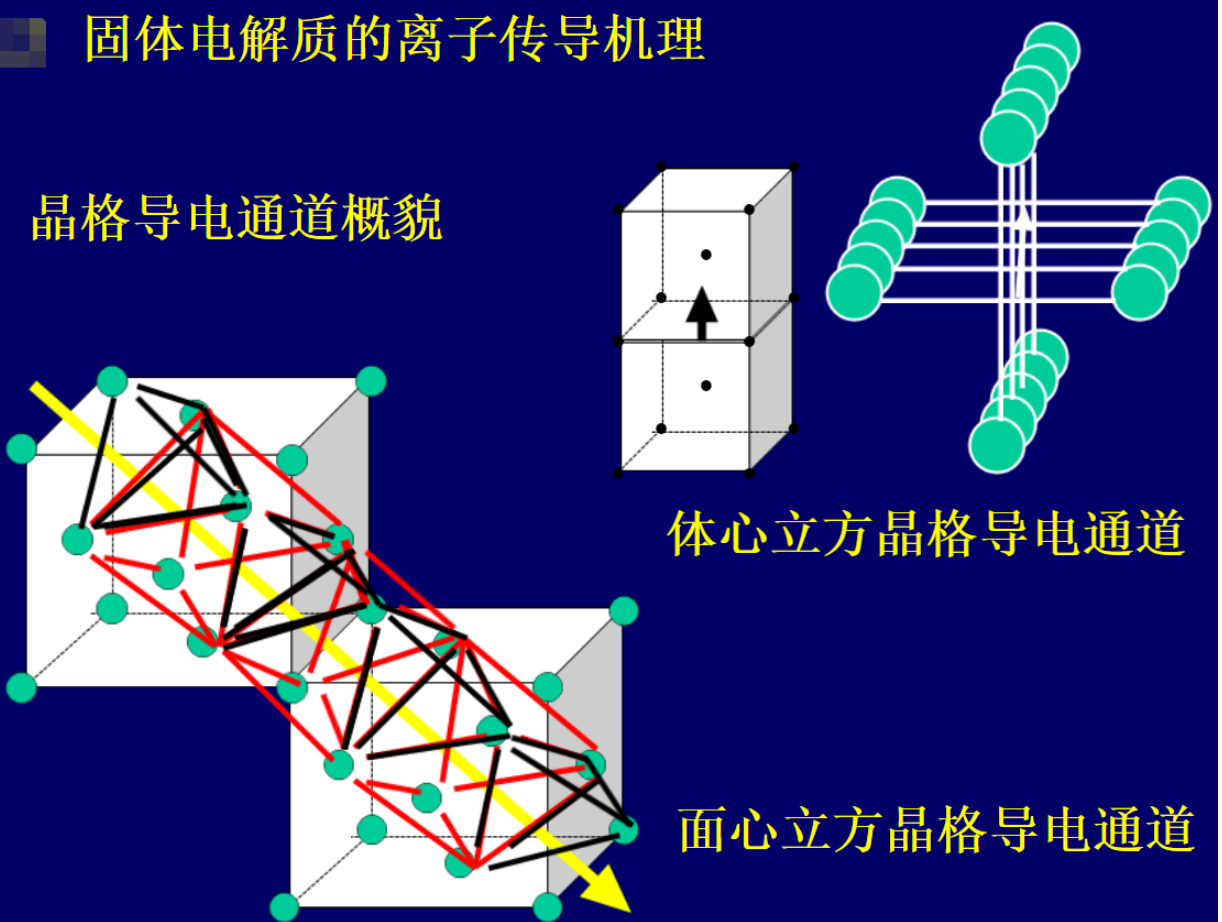
固態電解質離子傳輸機理解析
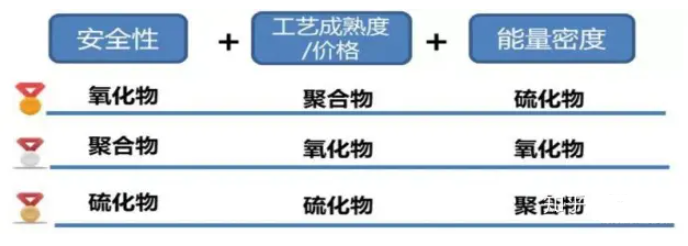

一種有機-無機非對稱固態電解質,實現長循環穩定的高壓鋰電池






 工商網監
工商網監
評論