 AEM綜述:硫化物基固態鋰電池的先進表征技術
AEM綜述:硫化物基固態鋰電池的先進表征技術
01
導讀
硫化物固體電解質具有非流動性、單離子導電性和低可燃性等優點,有望實現高能量和功率密度的下一代二次電池。然而,固體電解質的(電)化學分解、界面處的機械降解、鋰枝晶生長以及活性材料中鋰擴散緩慢等問題限制了其實際應用。
02
成果簡介
近日,Advanced Energy Materials上發表了一篇題為“Advanced Characterization Techniques for Sulfide-Based Solid-State Lithium Batteries”的綜述,對硫化固體電解質固態鋰電池的上述四個問題進行了綜述,還介紹了X射線光電子能譜、飛行時間二次離子質譜、透射電子顯微鏡和X射線計算機斷層掃描等先進表征技術在解決這些問題方面的現狀和前景。
03
關鍵創新
概述了硫化物基固態鋰電池的主要挑戰,以及硫化物基固態鋰電池先進表征技術的現狀和未來展望。
04
核心內容解讀
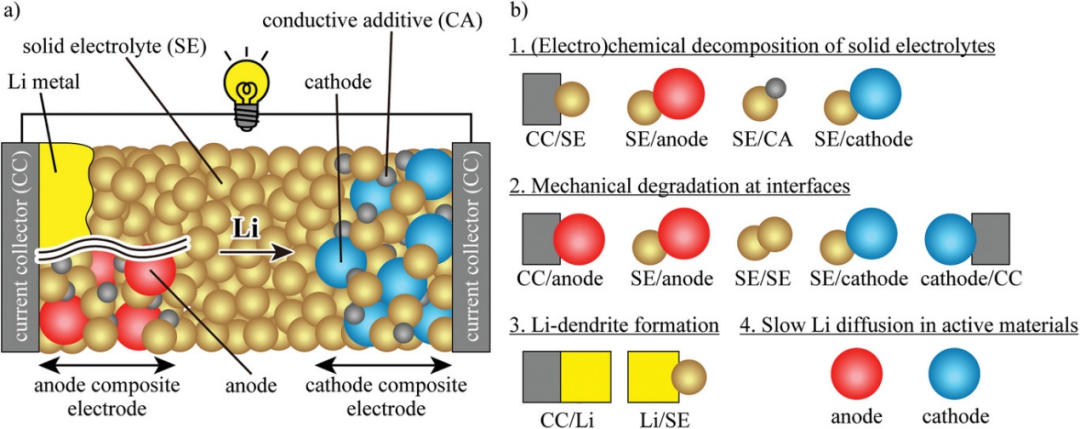
圖1. a)固態鋰電池構型。b)界面和活性物質中的主要問題。@Wiley
1、硫化物基固態鋰電池的問題
圖1a顯示了含硫化物SEs的固態鋰電池示意圖。負極是鋰金屬或復合電極結構。其中,存在各種固-固界面。這篇綜述主要關注四個根本問題,即硫化物SEs的(電)化學分解,界面的機械退化,Li枝晶形成,,活性物質中Li擴散緩慢(圖1b)。
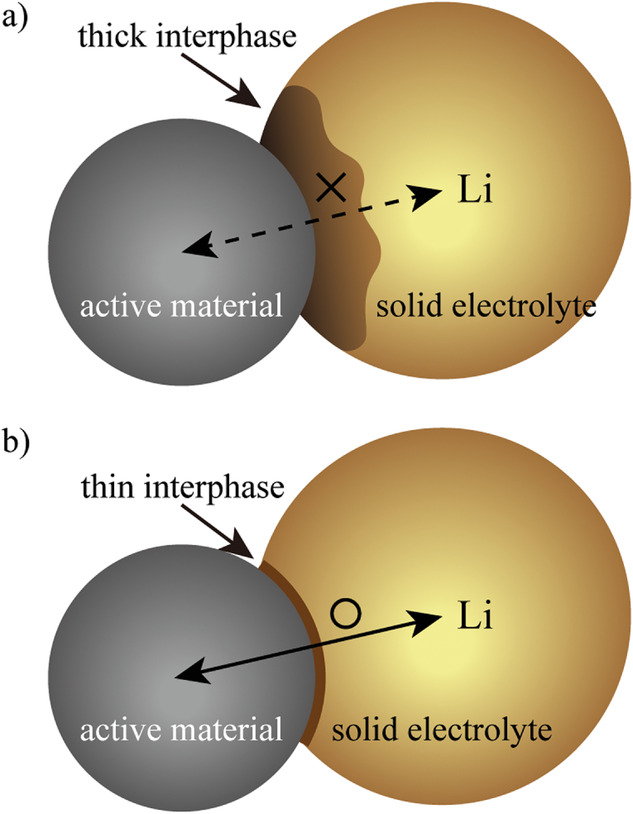
圖2. 活性物質和固體電解質(SEs)之間形成的界面示意圖。a)高電子導電性界面相。b)低電子導電性界面相。@Wiley
2、硫化物SEs的(電)化學不穩定性
在硫化物SEs中,(電)化學不穩定性是一個重要問題。在高于氧化穩定性的電位下,鋰從硫化物SEs中脫出,形成貧鋰的正極-電解質界面相(CEI)。在低于還原穩定性的電位下,隨著P5+或金屬元素的還原,Li被插入到硫化物SEs中,形成富Li的固體電解質界面相(SEI)。
界面相的電子電導率和離子電導率是決定界面電阻的關鍵。圖2a,b顯示,當界面相電子電導率較高時,分解后的界面相具有與活性物質相同的電勢,導致SE逐漸分解,界面相迅速增厚,阻礙了界面處的Li傳輸,降低了電池容量。相反,如果界面相是電子絕緣的,在活性物質的電位下,界面相將硫化物SE鈍化,從而抑制分解。
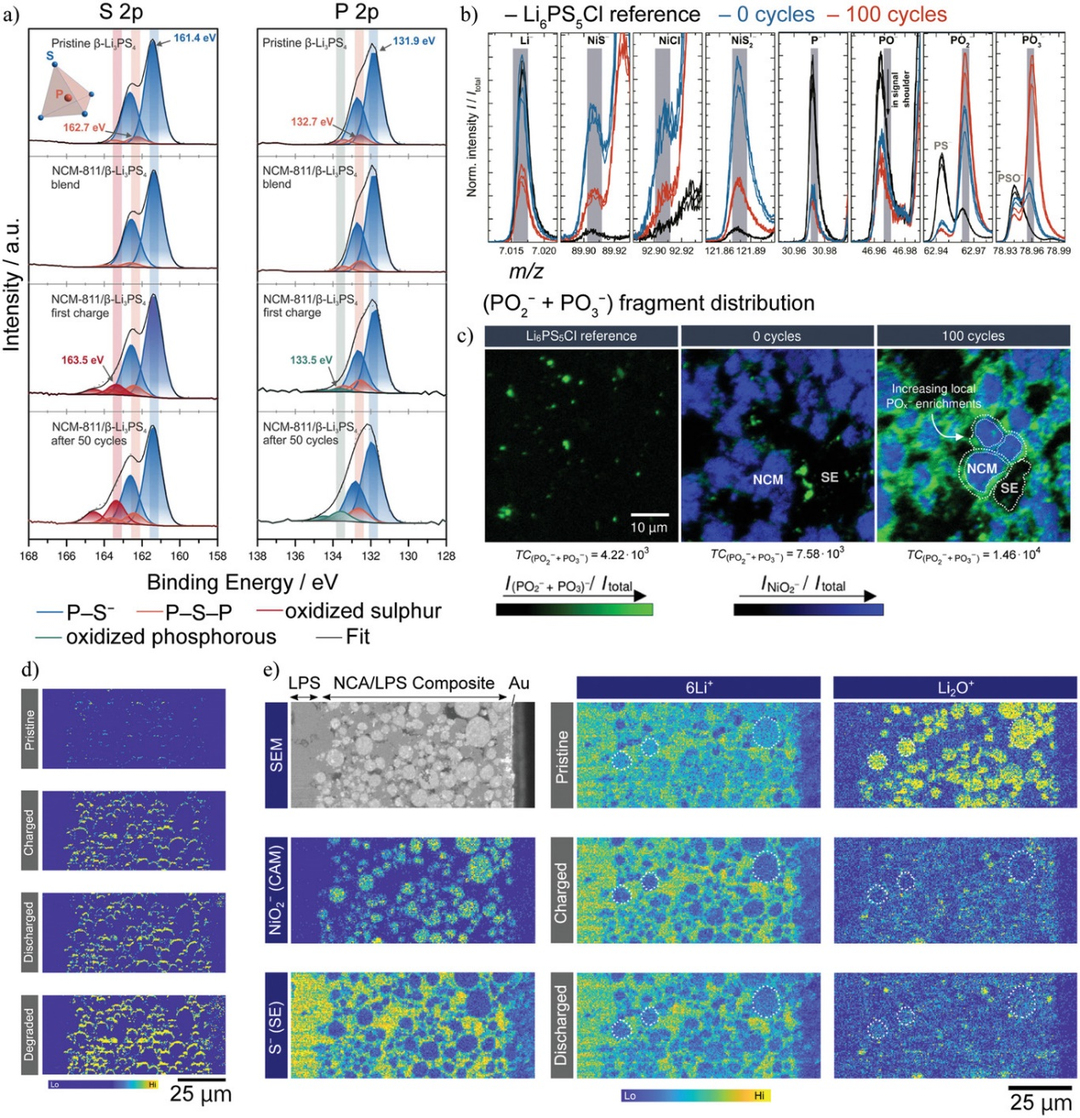
圖3. a)β-Li3PS4、LiNi0.8Co0.1Mn0.1O2(NCM-811)/β-Li3PS4復合正極初始狀態、第一次充電(0.1C)后、50次循環后的S-2p和P-2p XPS光譜。b)LiNi0.6Co0.2Mn0.2O2/Li6PS5Cl中帶負電的離子碎片的飛行時間二次離子質譜(ToF-SIMS)。c)NiO2?(藍色)和POx?(綠色)碎片的局部分布。d)通過原位ToF-SIMS測量LiNi0.80Co0.15Al0.05O2(NCA)/Li2S-P2S5微晶玻璃(LPS)復合正極中POx?碎片的演化。e)左:NCA/LPS復合正極的掃描電鏡(SEM)圖像和NiO2?和S?碎片的分布。中:6Li+碎片在充放電循環中分布的演變。右:Li2O+碎片在充放電循環中分布的演變。@Wiley
3、硫化物SEs的CEIs表征
通過XPS可以研究CEI的電子狀態。原始狀態下,只檢測到P-S?(161.4 eV)和P-S-P(162.7 eV)的XPS峰。在充電和循環狀態下,在高能量側觀察到一個寬峰,表明CEI含有帶有-S-S-鍵的多硫化物。該寬峰強度隨充放電反應而增大或減小,表明CEI隨電勢變化而發生氧化還原反應。XPS被廣泛用于表征界面反應,因為它可以測量SEs和CEIs中S和P電子狀態的微小差異。然而,XPS的空間分辨率較低,不能用于研究復合電極中CEIs的空間分布。
還可以使用亞微米空間分辨率的ToF-SIMS表征CEI。與XPS相比,ToF-SIMS的優點是具有更高的空間分辨率和化合物靈敏度。圖3b顯示了Li6PS5Cl、NCM-622/Li6PS5Cl電極在混合后以及100次循環后的碎片強度。100次循環后,樣品中的PO2?和PO3?碎片增加。同樣,觀察到SO2?和SO3?碎片增加。此外,利用ToF-SIMS的高空間分辨率,還能顯示正極附近CEI的空間分布。圖3c顯示,100次循環后,樣品中POx?碎片的強度在NCM-622附近增加,表明硫化物SE與NCM-622發生了反應。
NCA和LPS復合電極中PO2?和PO3?碎片的綜合強度分布結果表明,充放電后正極顆粒周圍的POx-碎片明顯增多。6Li+碎片代表NCA和LPS中的Li分布,而Li2O+碎片代表NCA中的Li分布。6Li+和Li2O+碎片強度在充電時下降,在放電時略有增加。然而,NCA中Li的濃度與碎片強度并沒有線性關系。盡管初始充放電效率為80%,但放電狀態下6Li+和Li2O+碎片的強度明顯低于原始狀態。層狀巖鹽正極的電子電導率在初始Li脫出過程中會增加,這可能會改變碎片的電離概率。另外,表面損傷也會導致放電狀態強度低。因此,定量分析顯然是ToF-SIMS中的一個問題。
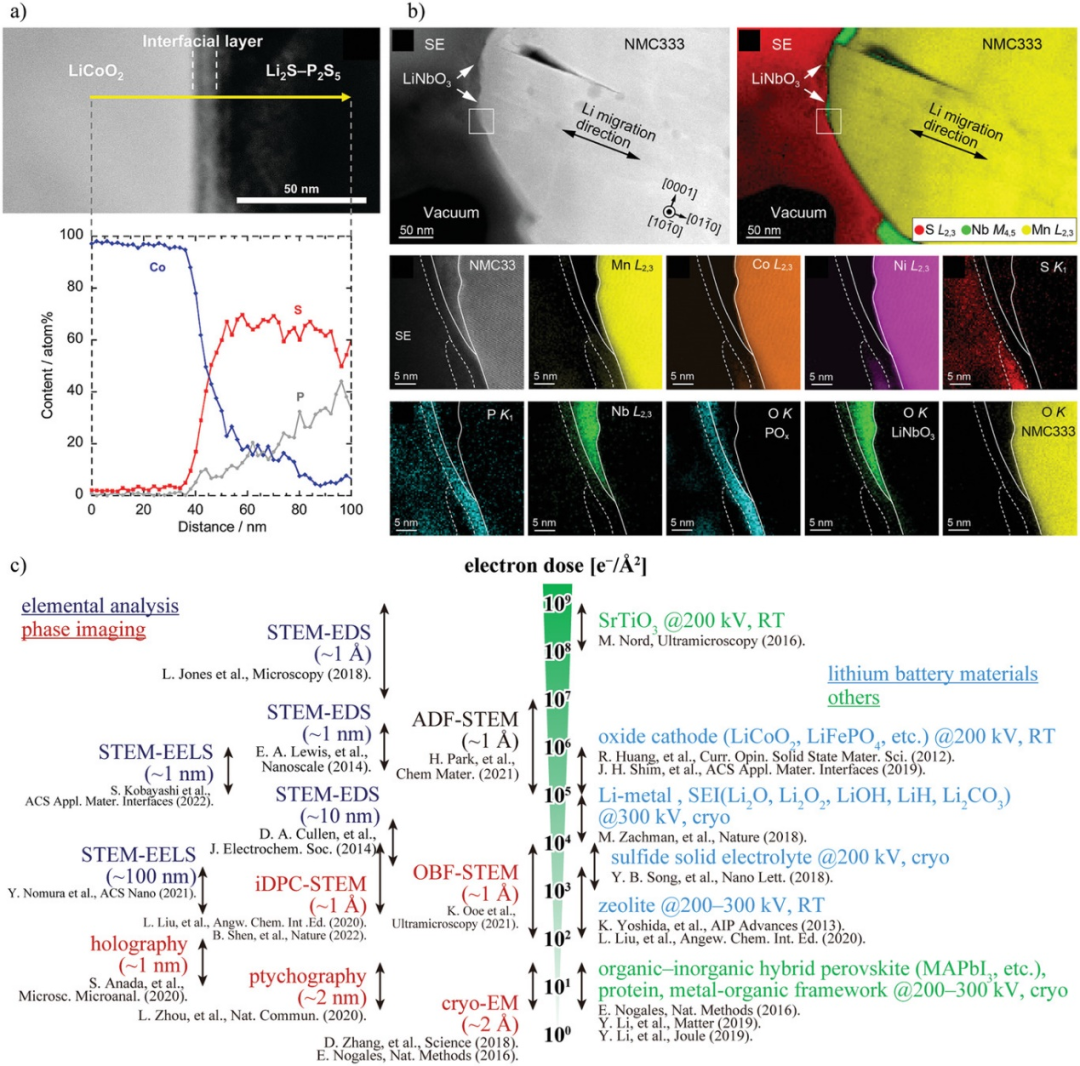
圖4. a)LiCoO2/LPS界面初始充電后的高角度環形暗場掃描TEM (HAADF-STEM)截面圖和Co、P、S的線掃描。b)通過電子能量損失譜(EELS)測量500個循環后LiNi1/3Mn1/3Ni1/3O2(NCM-333)/LPS界面的HAADF-STEM圖像和元素分布。c)電子束敏感材料TEM分析的總電子劑量。@Wiley
由于具有較高空間分辨率,TEM已被應用于正極與硫化物SEs界面的表征。在約10 nm的界面層中,Co、P和S相互擴散,表明Co擴散到LPS中阻礙了Li的傳導,增加了界面電阻。在NCM-333與LPS直接接觸的區域,Mn、Co、Ni向SE擴散。而在NCM-333被LiNbO3覆蓋的區域,過渡金屬沒有擴散,表明LiNbO3抑制了擴散。此外,在SE表面觀察到5 nm的POx層,在NCM-333表面觀察到5~10 nm的O耗盡層。
然而,硫化物SEs的TEM分析中存在電子束損傷問題。圖4c左側顯示了基于TEM的表征技術使用的單位面積電子劑量。圖4c右側顯示了觀察典型電子束敏感材料所用的單位面積電子劑量。紫色的元素分析(STEM-EELS和STEM-EDS)手段需要相對較大的電子劑量。對于空間分辨率≈1 ?的STEM-EDS,需要≈108 ??2的高電子劑量。因此,對于≈10 nm的空間分辨率,可以在≈104 ??2的劑量下進行STEM-EELS和STEM-EDS。考慮到鋰電池材料的電子束敏感性,LCO和LiFePO4等氧化物正極對電子束抵抗能力相對較強,在200 kV的加速電壓下,電子束劑量≈105-107 ??2。而鋰金屬,二元含鋰化合物,如Li2O, LiOH和Li2CO3,以及硫化物SEs對電子束更敏感。它們的結構在電子束劑量為103-105 ??2時降解。使用空間分辨率為≈1 nm(≈106 ??2)的STEM-EELS和STEM-EDS進行元素分析將導致SEs分解。
使用電子束相位信息的技術,如積分差分相位襯度(iDPC)STEM、最優明場STEM、疊層成像、電子全息和低溫電子顯微鏡(cryo-EM),在104 ?-2或更低的電子束劑量下,可以獲得小于1 nm的空間分辨率。然而,這些技術只能觀察原子排列或電位/電場分布,元素分布和電子狀態無法被表征。這些技術可以減少EELS分析的電子劑量,使正極/硫化物SE界面具有較高的空間分辨率和較低的損傷。
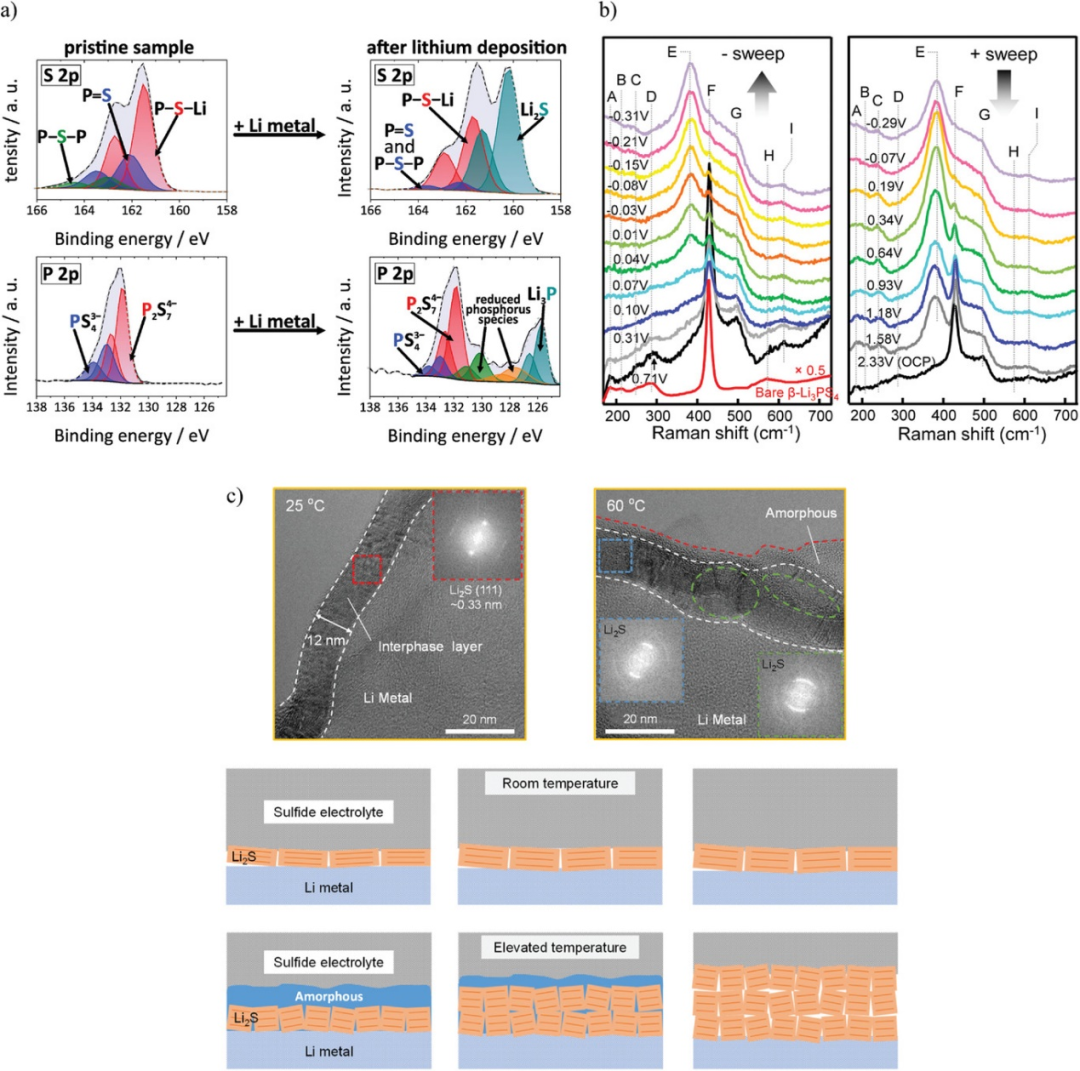
圖5. a)Li/Li7P3S11界面S-2p和P-2p光譜。b)鋰電鍍(左)和剝離(右)過程中β-LPS的原位拉曼光譜。c)Li/Li6PS5Cl界面處SEI的高分辨率低溫EM圖像。@Wiley
4、SEIs的表征
SEI最常用的表征技術是XPS。圖5a為Li7P3S11鍍鋰前后的S-2p和P-2p XPS譜圖。鍍鋰后,S-2p譜在160 eV處出現一個屬于Li2S的峰,P-2p譜在125.5 eV處出現一個屬于Li3P的峰。在Li電鍍過程中,歸因于拉曼光譜β-LPS中PS43?對稱拉伸的F峰強度隨著電位的變化而降低,而歸因于P2S64?中PS3對稱拉伸的E峰強度增加。結果表明,在鍍鋰過程中,β-LPS的還原作用使PS43?轉化為P2S64?。這表明PS43?和P2S64?之間的變化是一種與電位有關的氧化還原反應。PS43?和P2S64?之間的差異無法使用EELS輕易區分,因為該技術對β-LPS和P2S64?中存在的P-S鍵敏感。XPS和拉曼光譜可以用于表征SEI,因為它們可以分別測量微小的電子和結構差異。
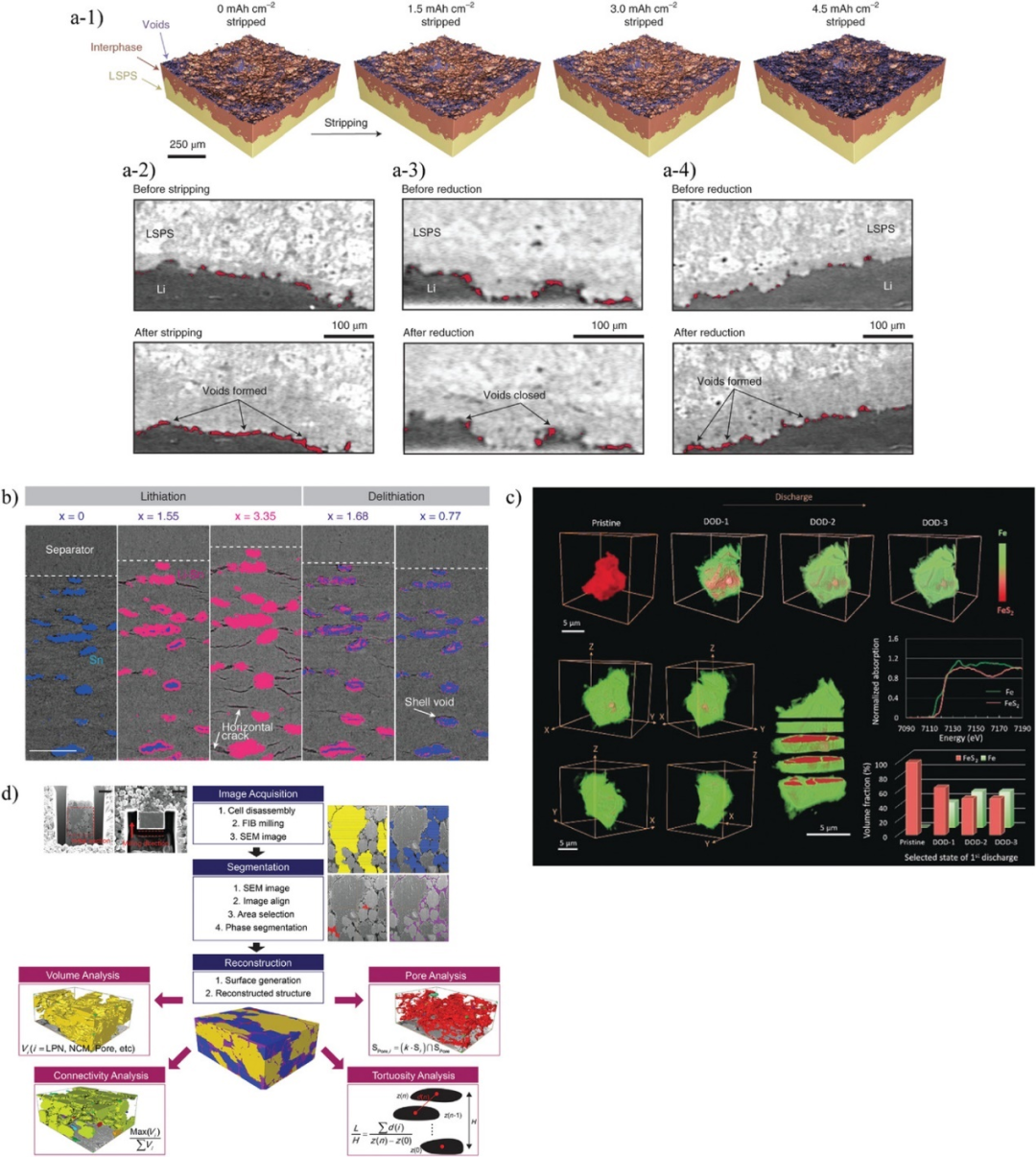
圖6. a-1)在1 mA cm?2的剝離過程中,通過原位CT觀察Li/LSPS/Li電池中Li/Li10SnP2S12(LSPS)界面上孔洞的演化。a-2)1 mA cm?2下Li/LSPS界面剝離前(上)和剝離后(下)的二維截面圖像。a-3)4 mA cm?2還原前(上)和還原后(下)的2D圖像。a-4)1 mA cm?2還原前(上)和還原后(下)的2D圖像。b)通過原位同步輻射X射線層析顯微鏡觀察Sn/LPS復合電極的結構演化。藍色和粉色區域分別表示Sn和LixSn。c)FeS2/LPS復合電極中FeS2和Fe的相演化隨放電狀態的變化。d)使用聚焦離子束和掃描電鏡三維重建技術進行定量分析的流程圖。@Wiley
5、界面的機械降解
由于活性材料的膨脹和收縮以及SEs分解引起的復合電極機械降解也是一個重要問題。它可能導致:1)離子或電子傳導通路彎曲度增加,2)活性材料/SE界面接觸面積減小,3)活性材料與SE分離,導致阻抗增加和容量衰減。
動態觀察包埋界面上的裂紋和接觸損失最常用的技術是X射線計算機斷層掃描(CT),它可以在毫米尺度上進行大面積的無損3D觀察。圖6a通過基于原位同步加速器的CT研究了在外加電壓下Li/Li10SnP2S12(LSPS)界面上產生的界面相和孔隙,并定量測量了接觸損失,表明界面接觸損失和重構是導致電池失效的關鍵因素。
圖6b利用同步輻射CT觀察了Sn/LPS復合電極充放電過程中Sn的體積變化和裂紋的形成。充電時,Sn吸收Li,轉變為LixSn。Sn在單軸加壓方向優先膨脹和收縮。裂紋在水平方向上形成,反映了Sn體積變化的各向異性。這些水平裂紋在析出后閉合,而Sn/LPS界面的界面分層始終存在。因此,CT能夠用于原位分析固態鋰電池的微觀結構和機械降解機制。
與CT相比,基于同步輻射的透射X射線顯微(TXM)技術具有更高的空間分辨率,可以用來研究界面處的局部現象。圖6c將原位TXM與X射線吸收近緣結構(XANES)相結合,以50 nm的空間分辨率觀察FeS2/LPS復合電極中FeS2在充放電過程中的化學狀態變化。通過分析Fe-K邊XANES光譜,可以發現FeS2向Fe轉變,形成核殼結構。在表面形成的Fe層能夠作為鈍化層阻礙Li的傳輸。TXM-XANES是一種能夠表征活性材料種過渡金屬價態變化的技術,其缺點是單次測量所需的時間較長。
圖6d使用FIB-SEM可視化了NCM-622/(Li2S)8(P2S5)2(Ni3S2)1(LPN)復合電極的三維結構。通過三維重構,可以量化各組分的體積比、連通性和彎曲度等結構信息以及這些組分之間的界面面積。結果發現,NCM-622/LPN復合電極包含15%的空隙,對電池性能產生了負面影響。另外,導電添加劑的分散性不佳,不能提供有效的電子傳導路徑。但這種方法存在相位分割問題。SEs、導電添加劑、粘結劑和空隙的掃描電鏡強度通常是相似的,因此很難準確分割。可以通過觀察掃描電鏡圖像并手動進行分割,但需要花費大量時間。此問題對FIB-SEM影響較大,但在CT和TXM中也很常見。
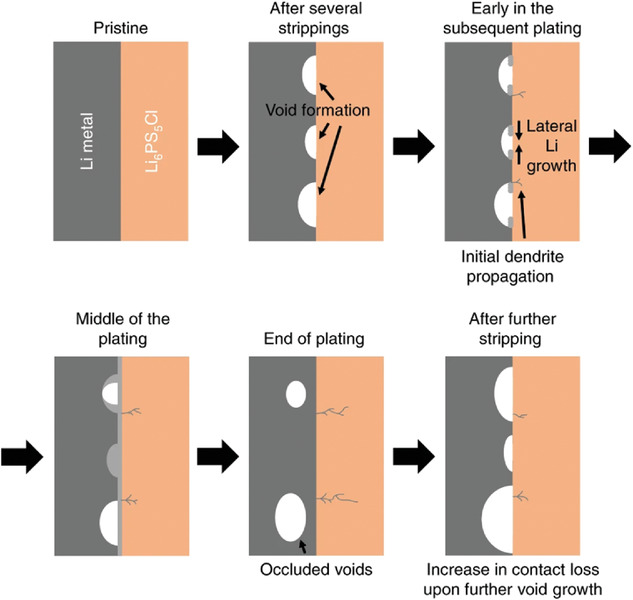
圖7. Li/Li6PS5Cl界面在高于臨界剝離電流的總電流密度下循環的原理圖。@Wiley
6、Li枝晶形成
硫化物SEs內部Li枝晶生長的原因之一是Li與硫化物SEs之間的粘附性較低,導致Li/SE界面電流分布不均勻。因此,Li在電流集中區域附近成核,導致枝晶生長。在鍍鋰過程中,CCD能夠判斷導致短路的最大電流密度。然而,在Li剝離過程中也具有CCD。圖7為大電流剝離時Li/Li6PS5Cl界面示意圖。首先,在界面附近的鋰金屬層產生孔洞。在隨后的循環中,Li沉積在孔隙中。然而,當孔洞非常大時,它們無法被填充,并且閉合的孔洞仍然停留在界面附近。隨著循環的進行,這些閉塞的孔隙增加,從而減少了接觸面積,增加了非均勻電流分布。這表明,當剝離電流較大時,電池更容易發生短路。

圖8. a)在與玻璃LPS斷口接觸的黃銅尖端電極上電鍍鋰的原位光學顯微鏡。b)多晶β-Li3PS4樣品斷口形貌的SEM圖像。@Wiley
光學顯微鏡是一種動態觀察晶體中枝晶和裂紋演變的技術。銅尖電極或SE表面均沉積有Li, SE表面未見枝晶生長。銅尖電極和SE表面均未見Li沉積。相反,在SE中觀察到新產生的裂紋輪廓。這表明,Li沿著表面缺陷生長并進入SE。多晶β-Li3PS4切割截面的SEM圖像,沿晶界或孔洞的Li枝晶為網狀。結果表明,表面缺陷和晶界是影響鋰枝晶生長的重要因素。光學顯微鏡的優點是可以原位地以高時間分辨率觀察Li電鍍過程。然而,這些僅適用于透明的SE。此外,在納米分辨率下進行3D分析和觀測并不容易。
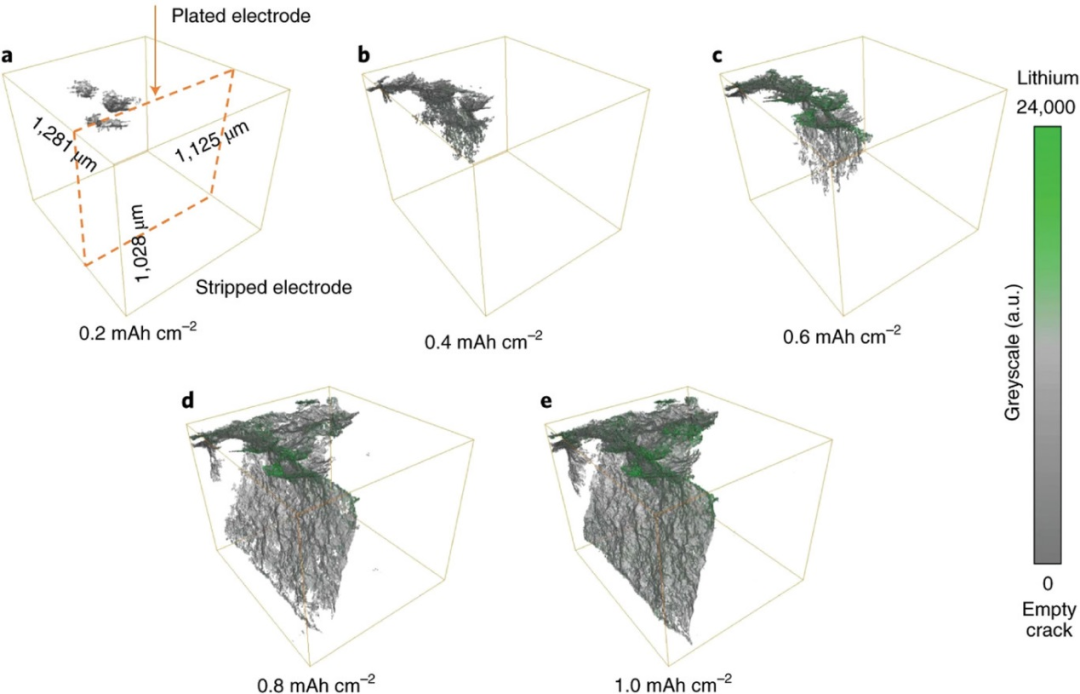
圖9. 在a)0.2 mAh cm?2、b)0.4 mAh cm?2、c)0.6 mAh cm?2、d)0.8 mAh cm?2和e)1.0 mAh cm?2電荷通過后,原位CT觀察到Li/Li6PS5Cl/Li對稱電池中Li枝晶形成。@Wiley
原位CT可以無損地顯示在SE層中Li枝晶生長的變化。具有高度相干光束的相位對比成像有效地增強了弱衰減材料的對比度,能夠將Li金屬與孔洞區分開來。這使得可以獨立地觀察裂紋擴展和鋰電鍍。在電池內部,SE裂紋首先在局部電場較大的Li電極邊緣形成。橫向裂紋沿SE層擴展到另一側的鋰金屬表面。圖9顯示了裂紋和鍍Li后裂紋內部沉積Li的原位CT的3D體積渲染圖像。當裂紋到達另一側Li金屬表面時,沒有發生短路,說明產生裂紋的整個區域沒有被鋰填充。鋰金屬通過從后部擴大裂紋,促進裂紋形成,隨后鋰金屬在裂紋內部生長,導致電池短路。
然而,由于CT的空間分辨率較低,很難分析局部結構,如裂紋尖端和枝晶形貌。而具有高空間分辨率以及三維分析能力的FIB-SEM只能觀察到一個狹窄的區域。要解決這一問題,可以將CT和FIB-SEM結合使用。首先,原位CT進行無損和3D成像,并確定感興趣的區域。隨后通過FIB-SEM對該區域進行分析,從而能夠將電化學性能與納米尺度下發生的局部現象聯系起來。
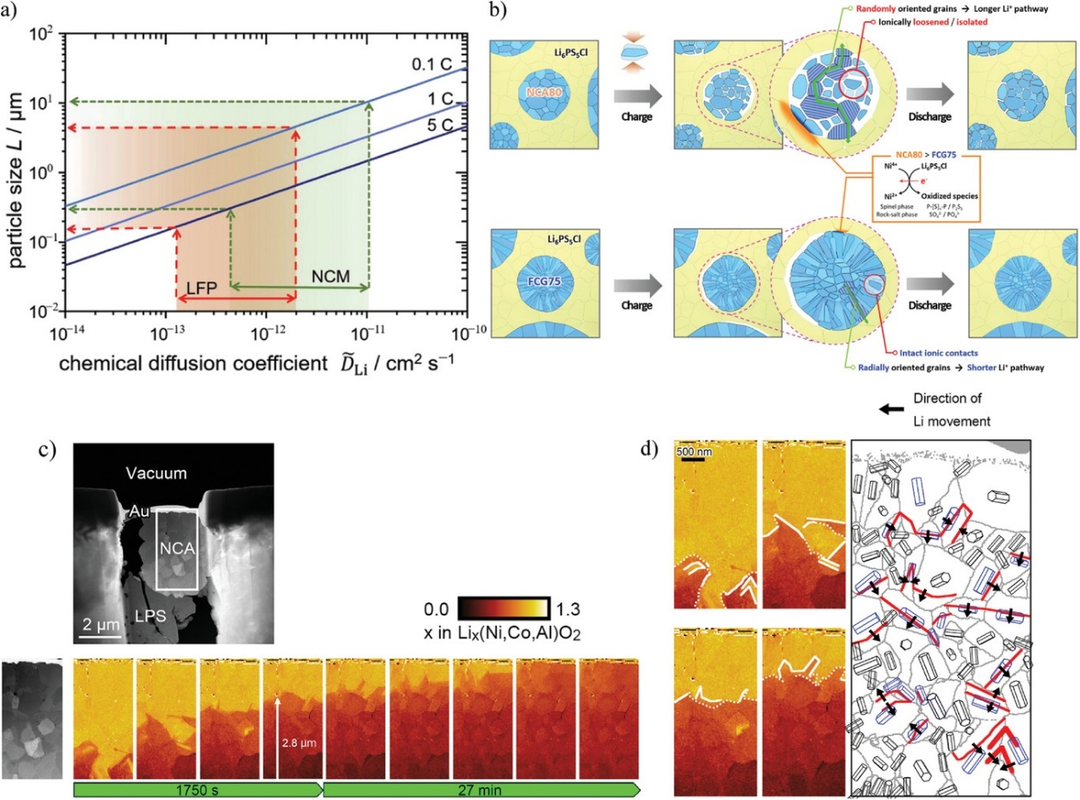
圖10. a)根據公式(1),當倍率為0.1,1,和5C時,取決于各自擴散系數范圍的最大粒徑。b)常規隨機取向正極(NCA80)和徑向對齊全濃度梯度正極(FCG75)示意圖。c)充電過程中NCA次級粒子中Li分布的原位STEM-EELS觀測。d)晶粒結構引導下的鋰離子運動。@Wiley
7、活性物質中緩慢的Li擴散
Li在活性物質中的擴散緩慢是另一個問題。如果活性物質中的Li擴散跟不上SEs的離子傳導,電化學反應就不能有效進行。粒徑較大的活性物質由于擴散長度較長,容易使輸出特性變差。
圖10a顯示了每種倍率下的化學擴散系數與粒徑之間的關系。從圖中可以看出,在1C下,NCM要達到83%的材料利用率,粒徑應小于幾微米。然而,如果活性物質粒徑非常小,則活性物質和SEs不能在復合電極內部高度分散。結果,與SE分離的活性物質比例增加,導致利用率降低。因此,在固態鋰電池中,鋰擴散對小顆粒尺寸的要求與離子傳導路徑對大顆粒尺寸的要求相沖突。要實現高功率運行和高活性材料利用率,必須促進活性材料內部的Li擴散。
優化粒子結構有望解決Li擴散問題。層狀巖鹽正極,如NCM,在晶格中有二維導Li平面(a-b面)。控制初級粒子的取向可以使鋰離子在徑向上快速擴散。圖10b顯示了常規隨機取向正極(LiNi0.8Co0.16Al0.04O2, NCA80)和全濃度梯度正極(LiNi0.75Co0.10Mn0.15O2, FCG75)的示意圖。二次粒子有望在徑向上實現快速的Li擴散,此外二次顆粒在徑向的體積變化較小,從而抑制了FCG75與SE界面處的機械降解。
對于這樣的粒子結構,研究Li擴散系數是很重要的。測定Li化學擴散系數最常用的方法是GITT和EIS。然而,這些方法只能獲得平均信息,而不能獲得局部信息。使用SIMS、原位Kelvin探針力顯微鏡(KPFM)和原位STEM-EELS能夠測量局部Li擴散。圖10c為NCA/LPS/InLi電池充電過程中NCA二次粒子內部Li濃度變化的原位STEM-EELS圖。Li從NCA中脫出,實現了對Li擴散的分析。圖10d顯示了Li擴散方向與晶體取向之間的關系。黑色箭頭所示的Li擴散方向在三方晶系NCA的基面(a-b平面),說明Li沿二維導電平面擴散。因此,原位STEM-EELS能夠以納米級的空間分辨率分析活性材料內部的局部Li擴散。
05
成果啟示
1)硫化物SEs與正負極都存在熱力學不穩定性問題。XPS、ToF-SIMS和拉曼光譜都有望揭示界面處的分解產物。電子顯微鏡能夠以更高的空間分辨率分析分解產物的晶體結構、確切組成和空間分布,但其存在電子輻照損傷。低劑量電子顯微鏡技術,如低溫電鏡,結合原位分析手段對于理解分解機制十分重要。
2)由活性物質體積變化和硫化物SEs分解引起的機械降解會破壞離子和電子傳導通路,導致阻抗增加和容量衰減。原位CT分析能夠動態觀察加壓條件下復合電極的三維結構,從而研究微結構與電池性能之間的關系。然而,CT的空間分辨率不足。原位CT或TXM結合具有更高空間分辨率的FIB-SEM有望提供更詳細的微觀信息。
3)鋰枝晶生長也是一個問題。利用原位表征技術,能夠可視化裂紋與枝晶擴展路徑。然而,這些因素之間的相關性還不完全清楚。使用具有高空間分辨率的原位三維分析方法將闡明這些問題。
4)鋰在活性材料中的緩慢擴散可能是限制硫化物基固態鋰電池功率密度的主要問題。通過設計特殊的晶粒結構有望促進鋰離子擴散,如全濃度梯度正極。原位KPFM和原位STEM-EELS可以在納米尺度上觀察活性材料內部的局部Li擴散。
審核編輯 :李倩
-
鋰電池
+關注
關注
259文章
8034瀏覽量
169528 -
電解質
+關注
關注
6文章
805瀏覽量
20017
原文標題:AEM綜述:硫化物基固態鋰電池的先進表征技術
文章出處:【微信號:Recycle-Li-Battery,微信公眾號:鋰電聯盟會長】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
全固態鋰電池領域的研究及實際應用
鋰電池VS聚合物鋰電池,誰才是未來的主角?
日本電池展上固態鋰電池大放光彩 將會成為動力電池的下一個風口
美國固態電池開發商Quantum Scape在紐約證券交易所上市

原位固態化聚合物電解質基高性能準固態軟包鋰電池
硫化物固態電解質與氧化物正極的熱穩定性


三菱綜合材料成功開發一種全固態鋰電池材料的制造新技術





 工商網監
工商網監
評論