 調控氟取代羧酸酯助力無負極電池
調控氟取代羧酸酯助力無負極電池
研究背景
在便攜式電子產品、電動汽車和電網存儲應用的方面,鋰離子電池(LIBs)需求正在大幅增長。然而,目前的LIBs中鋰過渡金屬氧化物正極(LiCoO2,LiMn2O4,LiFePO4等),與石墨負極(理論比容量為372mAhg?1),正接近能量密度的上限(~300 Whkg?1)。為了在實現電池水平上400 Whkg?1的能量密度,一個可行的選擇是使用鋰金屬負極(LMA)匹配高電壓,高比容量富鎳NCM(LiNixM1-xO2,M=Mn,Co,和x>0.6)正極。而電解質是實現高活性正負極的關鍵所在,電解質工程將改變LMA和NCM811正極的界面化學性質,因此被認為是實現高壓鋰金屬電池(LMB)的關鍵和可行的方案。
成果簡介
近日,中國科學院大學索鎏敏教授團隊提出了一種使用最佳的氟化線性羧酸酯(3,3,3-三氟丙酸乙酯,tFEP)與弱溶劑化氟碳酸乙烯酯(FEC)和解離鋰鹽(LiBF4和LiDFOB)來制備弱溶劑化解離電解液(WSDE)。在氟化線性羧酸酯中引入了–F來精確控制溶劑化能力并促進富LiF的界面,其中tFEP具有最佳的溶劑化能力和介電常數,在WSDE電解質中形成陰離子富集界面。Li-Cu庫倫效率(CE)高達98.7%,在4.6 V下匹配NCM811和LiCoO2正極穩定循環,且無負極軟包電池在惡劣的測試條件下可提供442.5 Wh kg?1的高比能量,100次循環后容量保持率達到80%。
研究亮點
(1) 引入–F取代來精確控制溶劑化能力并促進富LiF的界面,其中tFEP具有最佳的溶劑化能力和介電常數。
(2) 實現在正負極表面形成穩定的EEI。
(3) 在高正極負載4.64 mAh cm?2,無負極,貧電解液 2.75 g Ah-1條件下裝配軟包電池,100圈容量保持率為80%。
(4) 上述無負極軟包電池能量密度達442.5 Wh kg?1。
圖文導讀
想要了解陰離子富集界面如何決定界面化學,首先要了解亥姆霍茲內平面(IHP)的結構,該結構是吸附離子電中心的軌跡,與電極|電解質界面(EEI)的形成密切相關。在WSDE電解質中,由電場驅動,Li+伴隨著初級溶劑化殼(CIP和AGG)中的陰離子,從本體電解質傳輸到電極表面的IHP(圖1b),IHP中的陰離子容易分解形成陰離子衍生的致密EEI(圖1a)并避免溶劑與電極直接接觸。此外,在WSDE電解質中,陰離子的還原電位也因與Li+的相互作用而改變。高度的Li+-陰離子締合有助于形成陰離子衍生的EEI(圖1c)。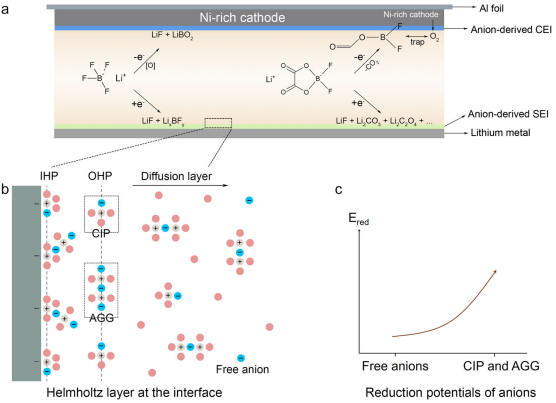
圖1. 富陰離子界面的形成原理。
當引入-F基團時,具有較高介電常數(1.87)的2-單氟丙酸乙酯(mFEP)與LMA不相容(圖2a)。將-F基團數量增加到5將使五氟丙酸乙酯(pFEP)具有極低的介電常數,不能溶解足夠的鋰鹽來保證電解質的離子電導率(圖2c)。而具有3-F取代基的3,3,3-三氟丙酸乙酯(tFEP)具有適當的介電常數1.79,與Li金屬相容,同時具有足夠的離子電導率。利用量子化學計算得到鋰離子與溶劑之間的結合能,以確定介電常數,其中結合能按mFEP > tFEP > pFEP的順序下降(圖2b)。
因此,由于最佳的介電常數、溶劑化功率和氟取代基,以及足夠的離子電導率,tFEP是本工作的主要溶劑。本研究將具有最佳介電常數和溶劑化能力的tFEP和FEC雙溶劑與弱解離的LiBF4和LiDFOB結合制備WSDE電解質(1 M LiBF4+1 MtFEP/LiDFOB),并加入FEC以提高離子電導率(圖2c)。采用核磁共振(NMR)技術探測電解質的離子-溶劑相互作用。11B NMR中代表BF4-的化學位移顯示紅移,且EC/DMC>mFEP>tFEP>pFEP(圖2d),表明BF4-與Li+的親和力越來越強,19F NMR具有實類似的化學位移趨勢(圖2e)。通過分子動力學(MD)模擬計算RDFs和CDFs,求解后Li+的微觀結構(圖2i-k)。
RDF表明,溶劑(FEC和FEP)和陰離子(BF4-和DFOB?)都涉及Li+初級溶劑化鞘的形成。DFOB?陰離子強烈地聚集在Li+周圍,第一個峰位置約為1.85 ??,而BF4-陰離子離Li+最遠,第一個RDF峰為3.15 ??。MD模擬結果與表征結果一致,證實了在1M LiBF4 + 1 M LiDFOB tFEP/FEC電解質中具有富陰離子的溶劑化結構。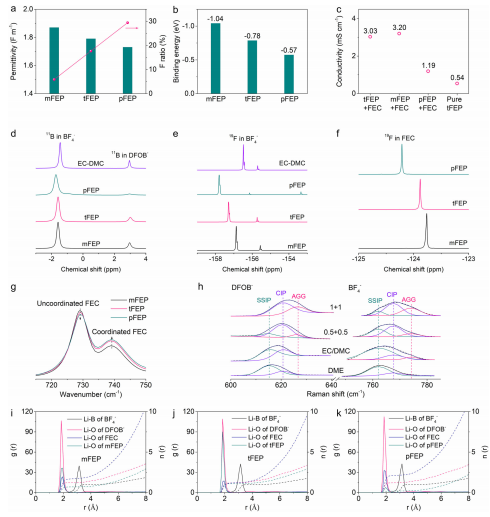
圖2. 溶劑化結構的演化和計算模擬。
為了揭示溶劑化結構對界面化學的影響,利用Li/Cu半電池研究了LMA的電化學性能。1 M LiBF4 + 1 M LiDFOB tFEP/FEC電解質在100個循環內提供鋰剝離/電鍍的平均CE為98.7%,遠高于mFEP基電解質(低于96%)和1M LiPF6 EC/DMC基礎電解質(~89%)(圖3a)。
相比之下,基于pFEP的電解質不能支持穩定的Li循環,可能是由于其低電導率為1.19m Scm?1(圖2c)。為了突出LiBF4 + LiDFOB雙鹽的優點,本工作在tFEP/FEC溶劑中使用各種鹽制備了電解質。在100個循環中,tFEP/CEP/FEC電解質中使用LiBF4的CE緩慢上升到~97.2%,而使用LiDFOB和LiPF6為基礎的電解質難以忍受長時間的循環(圖3b)。
在1M LiBF4 + 1 M LiDFOB tFEP/FEC電解液中,鋰無縫沉積在銅集流體上,形成完美的柱狀結構,孔可忽略,厚度為29.32μm(圖3d)。在1M LiPF6 EC/ DMC電解質中,多孔鋰沉積物以不受控制的方式生長,厚度為54.75 μm。弱鍵導致Li沉積物與Cu底物之間出現明顯的裂紋(圖3d),使得部分Li沉積物從Cu底物中剝離。
從TOF-SIMS(圖3f)和XPS(圖3g,h)的結果來看,與1M LiPF6 EC/DMC基線電解質中SEI的鑲嵌模型不同,1M LiBF4 + 1 M LiDFOB tFEP/FEC電解質中的SEI主要由無機組分組成。緊湊而堅固的SEI具有高楊氏模量,可以有效抑制鋰樹突的生長,從而提高安全性和壽命。
此外,無機豐富的SEI會產生豐富的相邊界和空位,促進Li+的擴散,降低間相過程的活化能。鋰離子通過SEI的均勻和快速擴散將調整致密和緊密排列的鋰沉積物的形成。堅固和快速的Li+導電SEI和因此密封的鋰床共同保證了LMA的高CE和長壽命。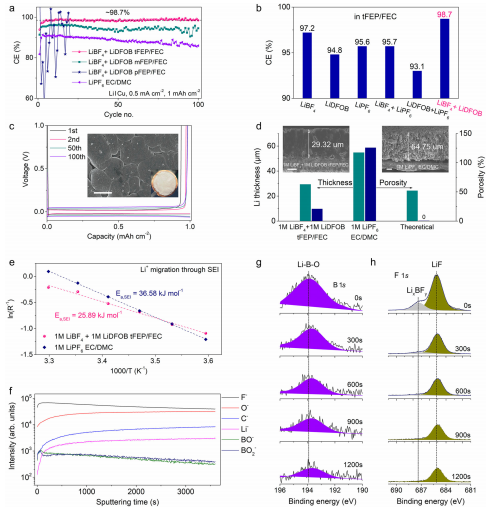
圖3. LMA的電化學性能和表征。
使用高活性4.6 V NCM811和LiCoO2正極評估了1M LiBF4 + 1 M LiDFOB tFEP/FEC電解質與高壓正極的兼容性(圖4)。當NCM811正極充電到4.6 V時,在0.2 C下提供224mAhg?1(圖4a)。在100個循環后的容量保持率為80.5%,CEs接近~99.6%。相比之下,在1M LiPF6 EC/DMC基線電解質中,只有52.9%的容量保持率,CEs小于99%。
此外,與基礎電解質相比,本電解質表現出更穩定的電壓分布和更高的能源效率(圖4b)。放電過程開始時的大電壓降主要可以歸因于NCM811正極退化引起的過電位和在循環過程中鋰金屬負極的SEI厚度的增加。 除了NCM811正極,本電解質與4.6 V LiCoO2正極同樣具有較好兼容。
在該電解質中,100次循環后容量205.6 mAhg?1,沒有明顯的電壓衰減(圖4c,d)。相比之下,在1M LiPF6 EC/DMC基礎電解質中,只有61.3%的容量保持率(圖4c)。通過靜電流間歇滴定技術(GITT)測量研究其基本原理(圖4e)。盡管在第一個周期有類似的過電位,100個周期后基礎電解質的過電位遠高于該電解質中,這可能是由于相變、結構損傷和電阻更高的相間層。使用電化學阻抗譜(EIS)對3電極電池進行測量,以排除LMA的影響(圖4f)。經過100次循環后,該電解質中CEI(高頻半圓)和電荷轉移過程(中頻半圓)的電阻遠低于基礎電解質。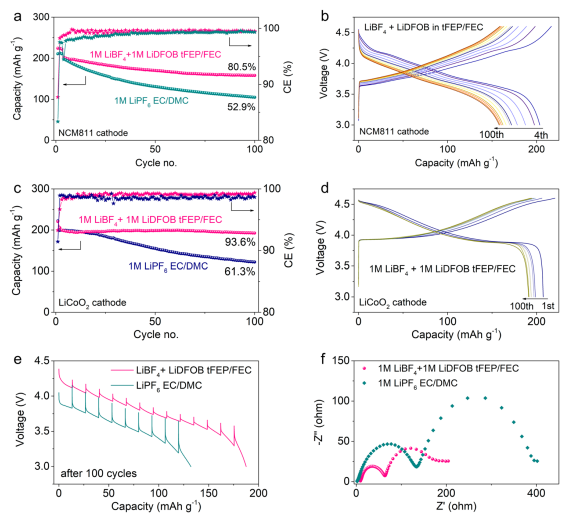
圖 4. 匹配4.6 V NCM811和LiCoO2正極的電化學性能。
與NCM811性能密切相關的正極界面化學性質得到了廣泛的研究(圖5)。首先在4.6 V下持續暴露正極和電解液之間的副反應,其中泄漏電流為副反應速率(圖5a)。在10,000秒保持時,基線電解質中的泄漏電流緩慢下降到4.6 μA mg?1。
相比之下,本電解液中的泄漏電流迅速衰減到0.2 μA mg?1的最小值。采用電感耦合等離子體質譜(ICP-MS)測量方法,研究了過渡金屬(TMs)的溶解度在正極降解過程中起著至關重要的作用。經過100個循環后,與基礎電解質相比,該電解質中溶解的TMs明顯較少(圖5b)。用高分辨率透射電鏡(HRTEM)直接觀察循環NCM811的CEI。在本電解液中循環的NCM811被4 nm厚的非晶CEI包裹;相比之下,基礎電解質中的正極界面表現出明顯的表面降解,很容易觀察到超過10 nm的巖鹽層(圖5f)。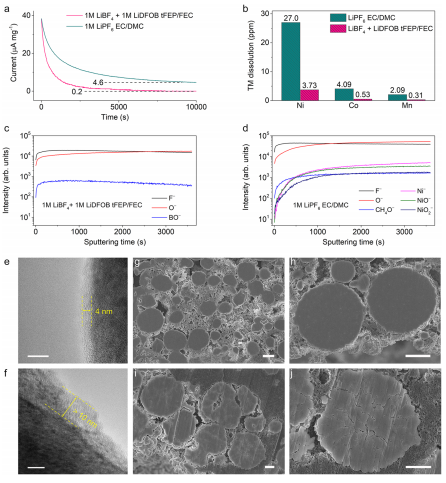
圖5. 在對比電解質中,4.6 V截止電壓下的正極化學表征。
為了系統地強調本工作電解液與LMA和4.6 V NCM811正極的兼容性,本工作在零過量鋰、高正極負載4.64 mAh cm?2、貧電解液2.5g Ah-1的惡劣條件下,構建了無負極軟包電池。本工作制備的雙層軟包電池容量為220 mAh,對應的能量密度為365.9Wh kg-1,在優化電解質中,在0.1C充電和0.5C放電倍率下進行100次循環后,容量保持率達80%(圖6a,b)。
而在基礎電解質中,袋電池只能存活20個周期,在此周期中,不受控制的副反應會迅速耗盡Li和/或電解質,并導致災難性的容量衰減(圖6a,c)。通過計算,無負極軟包電池的電池水平比能量為442.5 Whkg?1,這在報道的LMBs中具有很高的競爭力。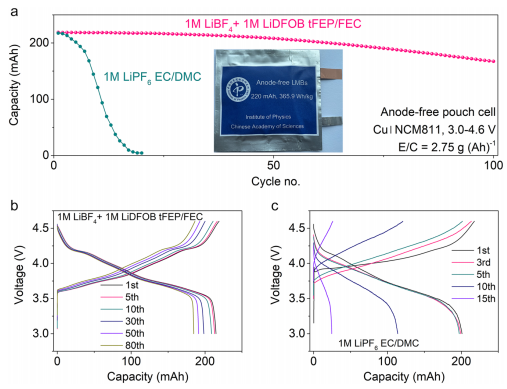
圖5. 無負極Cu|NCM811軟包電池的電化學性能。
總結與展望
在本工作設計的弱溶劑化和解離電解質中,通過引入氟化線性羧酯與鋰鹽(LiBF4和LiDFOB)結合弱解離,獲得最佳的溶劑富集界面。陰離子富集界面導致亥姆荷茲平面內的陰離子分解更多,陰離子的還原電位更高,有助于無機豐富的界面化學。
因此,得到了致密柱狀鋰沉積,~CE為98.7%。4.6 V NCM811和LiCoO2正極的穩定循環可以超過100個循環,邊際電壓衰減,得益于被抑制的正極-電解質副反應、NCM二次粒子的裂解和TM溶解。因此,構建了工業無負極軟包電池(>200 mAh),在高正極負載4.64 mAh cm?2)和貧電解質(2.75 g Ah?1)的惡劣測試條件下,100次循環后容量保持率80%,且對應于估計的能量密度為442.5 Wh kg-1。本工作提出的稀電解質中陰離子富集界面的方法將為鋰金屬高壓富鎳電池的精確工程提供指導。
審核編輯:劉清
-
鋰離子電池
+關注
關注
85文章
3215瀏覽量
77544 -
電解質
+關注
關注
6文章
804瀏覽量
20017 -
FEC
+關注
關注
0文章
40瀏覽量
13676 -
便攜式電子
+關注
關注
0文章
5瀏覽量
9242
原文標題:索鎏敏最新Nature子刊—調控氟取代羧酸酯助力無負極電池攀登400 Wh kg-1高峰
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦

石墨負極在鋰離子電池中的發展與儲鋰機制
鐵電池與磷酸鐵鋰電池區別是什么
Haydale血糖和甘油三酯電化學監測導電油墨
鎳氫電池的負極材料是什么

氟離子電池中涉及O?O鍵形成的高容量雙層鈣鈦礦氟氧化物正極


鋰電池負極材料研磨常用設備

5號電池正負極的區分方法

高比能無負極軟包電池,未來多遠?






 工商網監
工商網監
評論