 細胞核結構電解液助力低溫水系鋅電池
細胞核結構電解液助力低溫水系鋅電池
研究背景
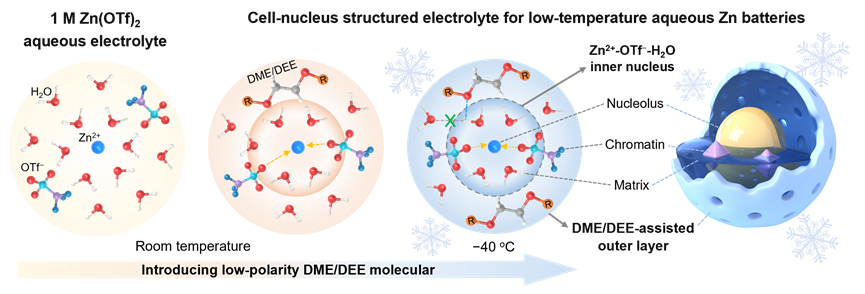
可充水系鋅電池(RAZBs)由于鋅負極的高理論容量、低氧化還原電位、無毒性以及易于制備與儲存特點,賦予該體系低成本、高容量、安全環保等優勢,在大規模儲能方面顯示出很大的應用前景。然而,最先進的RAZBs仍受到鋅負極在傳統水系電解液中可逆性差、枝晶生長、析氫和表面腐蝕等問題,這在高放電深度(DOD)下更為嚴重。除此之外,由于水系電解液熱力學冰點高、低溫反應動力學緩慢,導致零度以下鋅電池的可逆容量大幅降低、循環壽命急劇縮短,嚴重限制了RAZBs低溫下的實際應用。因此,抑制水系電解液低溫凍結、提升鋅負極的可逆性與穩定性,成為解決上述問題的理想方案。
溶劑化結構在調節界面化學、鋅負極可逆性和電池性能方面發揮著至關重要的作用。然而,高鹽濃度和高極性有機溶劑的引入不可避免地增加了Zn2+的去溶劑化能,降低電解液的離子電導率和Zn2+擴散動力學,導致RAZBs在低溫條件下的電化學性能令人不滿。鑒于此,南開大學程方益教授和河北大學張寧教授課題組通過設計一種獨特的“細胞核”型溶劑化電解液結構,實現了水系鋅離子電解液在超低溫下保持液態,并獲得了優異的?40 ℃低溫鋅電池性能。同時,本工作為低溫水系電解液的溶劑化結構設計提供了新的思路。該工作以“Cell-nucleus structured electrolyte forlow-temperature aqueous zinc batteries”為題發表在Journal of Energy Chemistry上。本文第一作者為南開大學博士后董陽,通訊作者為程方益教授和張寧教授,第一通訊單位為南開大學化學學院。
研究亮點
引入低極性鏈醚類溶劑構建新型“細胞核”型溶劑化結構,打破水系電解液中原始氫鍵網絡,擴大液態溫度范圍;
富含陰離子的Zn2+-PSS(內核)和有機溶劑調制的Zn2+-外溶劑化鞘(外層)結構,促進低溫下Zn2+的遷移擴散與去溶劑化動力學;
促進室溫及低溫下在鋅負極表面原位形成有效SEI,有效抑制鋅枝晶和寄生反應;
實現?40 ℃下Zn||Cu、Zn||Zn和Zn||V2O5電池的高倍率穩定工作。
圖文導讀
圖1. 電解液耐低溫性質測試.
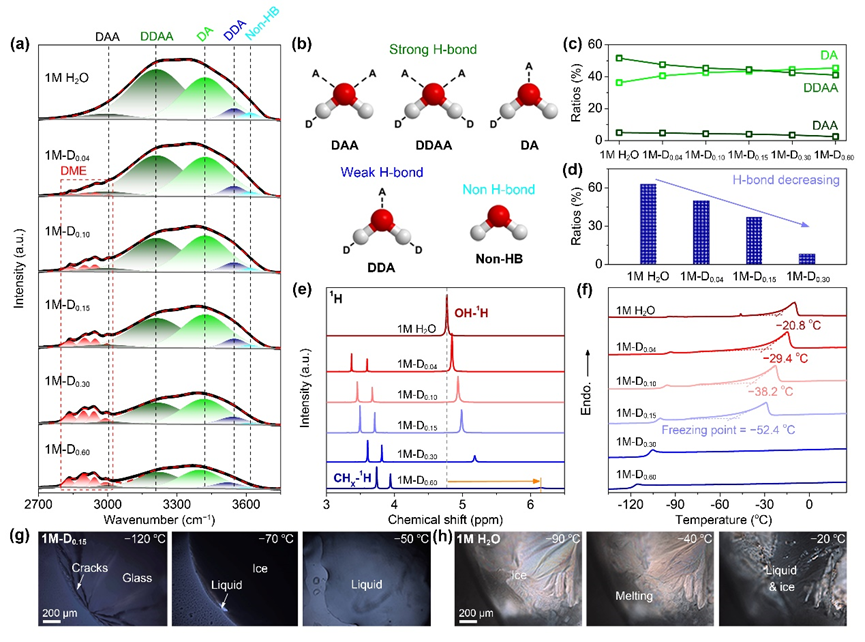
(a) 1M-Dx電解液的FTIR光譜;(b)DAA、DDAA、DA、DDA和Non-HB的示意圖模型;(c)強氫鍵水分子的比例演變;(d) MD模擬水間原始氫鍵比例;(e) 1H NMR譜;(f)?150 ~ 25 °C范圍內電解液的DSC曲線。(g) 1M-D0.15和(h)1M H2O在不同溫度下的偏光顯微鏡觀察圖像。
▲通過紅外核磁等譜學測試分析了電解液體系中水氫鍵的破壞與重整。根據水分子間的通過不同電子供體/受體的H/O原子的配位情況,將氫鍵分為(b)圖中的5種類型。隨著DME添加量增多,強氫鍵中DAA和DDAA占比降低,而同時DA的比例逐漸增多,這表明水中強氫鍵網絡的破壞和松動,將有利于降低電解液的低溫凝固點并保持水分子快速的振動/旋轉動力學,提高低溫電導率。圖(d)中MD模擬計算表明引入DME后水間原始氫鍵明顯減少。水中1H-NMR信號的藍移,H周圍電子云密度增加,也說明了水中原始氫鍵的破壞。利用DSC測試了電解液的低溫凝固點,結果表明最優1M-D0.15體系的冰點低至?52.4 oC。進一步利用偏光顯微鏡表征分析了電解液隨溫度降低的結晶相變行為。相比于1M H2O在-20度下水的成核結晶,1M-D0.15直到?70 oC才出現冰核,?120 oC發生玻璃化轉變,這與DSC結果保持一致。
圖2.溶劑化結構表征.
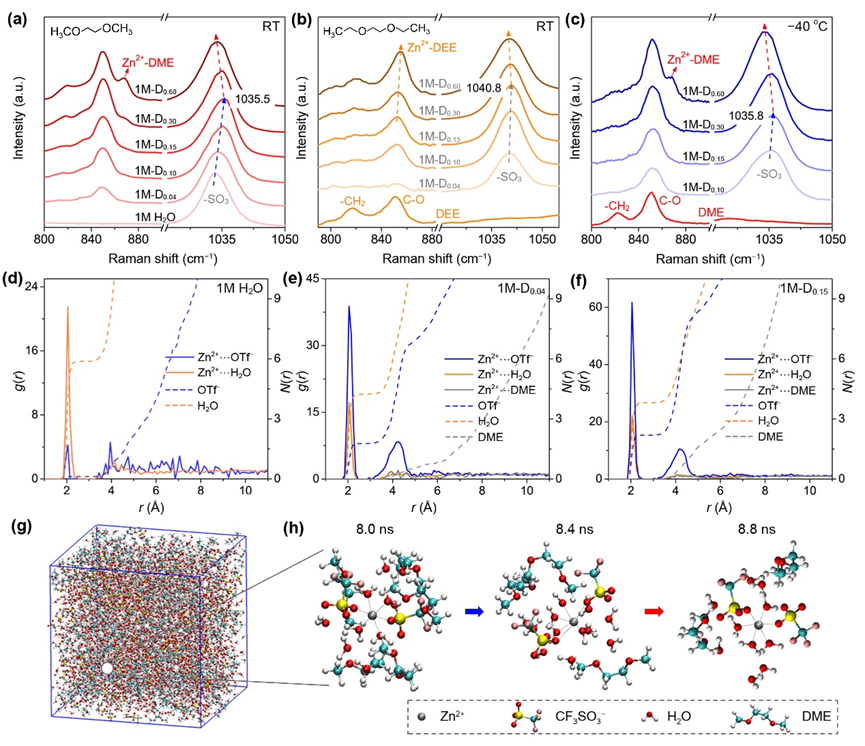
(a)DME和(b)DEE輔助電解液在室溫下800 ~ 880 cm?1(DME/DEE的?CH2搖擺振動和C?O伸縮振動)和1020 ~ 1050 cm?1(OTf?的SO3伸縮振動)范圍內的拉曼光譜;(c)不同DME電解液在?40 ℃的低溫拉曼光譜;(d) 1M H2O、(e) 1M-D0.04和(f) 1M-D0.15電解液的RDF g(r)和配位數N(r)圖;(g) 1M-D0.15的快照和(h) 8.0、8.4、8.8 ns時Zn2+的雙層溶劑化結構的構型演變。
▲利用原位變溫拉曼光譜表征室溫和低溫下DME和DEE系列電解液,發現當添加劑的摩爾占比小于0.3時,DME/DEE不會與Zn2+直接配位,并且隨著醚溶劑的添加,Zn2+與OTf?間相互作用逐漸增強。但添加更多時,醚分子開始參與到Zn2+的第一溶劑化鞘層中,受空間位阻等的影響,導致陰陽離子間的配位作用降低。DME體系在?40 oC低溫下的測試中也呈現了相似的趨勢。并結合MD模擬對比了純鋅鹽水溶液和小劑量DME添加的電解液中鋅離子的徑向分布函數和配位數,與上述拉曼測試結果相符。并通過隨機截取最優體系的瞬時模擬快照,驗證了目標“細胞核”型Zn2+雙溶劑化鞘層結構的形成。
圖3.鋅負極性能測試.
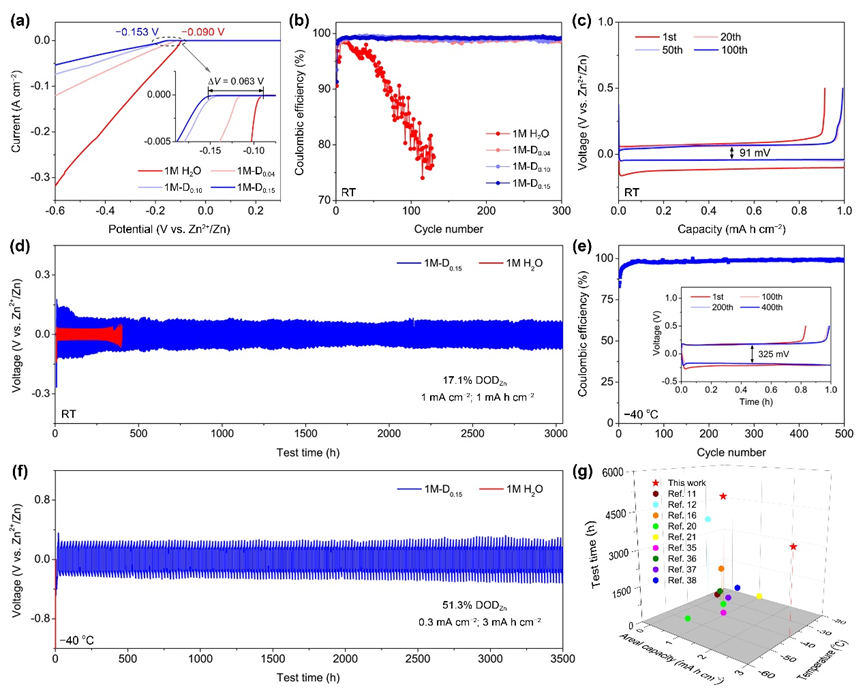
(a)不同電解液的析氫極化曲線;(b)室溫下Zn||Cu電池的鍍鋅/剝離CE,(c)1M-D0.15對應的電壓-容量曲線;(d)室溫下Zn||Zn電池在1M-D0.15和1M H2O電解液中的恒電流循環性能;?40 ℃下(e)Zn|1M-D0.15|Cu半電池的沉積/析出CE和電壓曲線(插圖)和(f)Zn||Zn電池的低溫循環性能;(g)本工作與最近報道低溫電解液的Zn||Zn電池性能對比圖。
▲采用LSV測試表明細胞核溶劑化結構有效推低了水系電解液的析氫電位。通過Zn||Cu半電池測試了電解液中鋅沉積/析出的可逆性,結果顯示我們設計的細胞核型電解液在室溫和?40 oC低溫下CE均可達到99.2%以上。并能夠支持Zn||Zn對稱電池在室溫和低溫下實現3000小時以上的超長穩定循環,與最近報道的低溫水系電解液相比顯示出明顯的優勢。
圖4.界面行為表征.
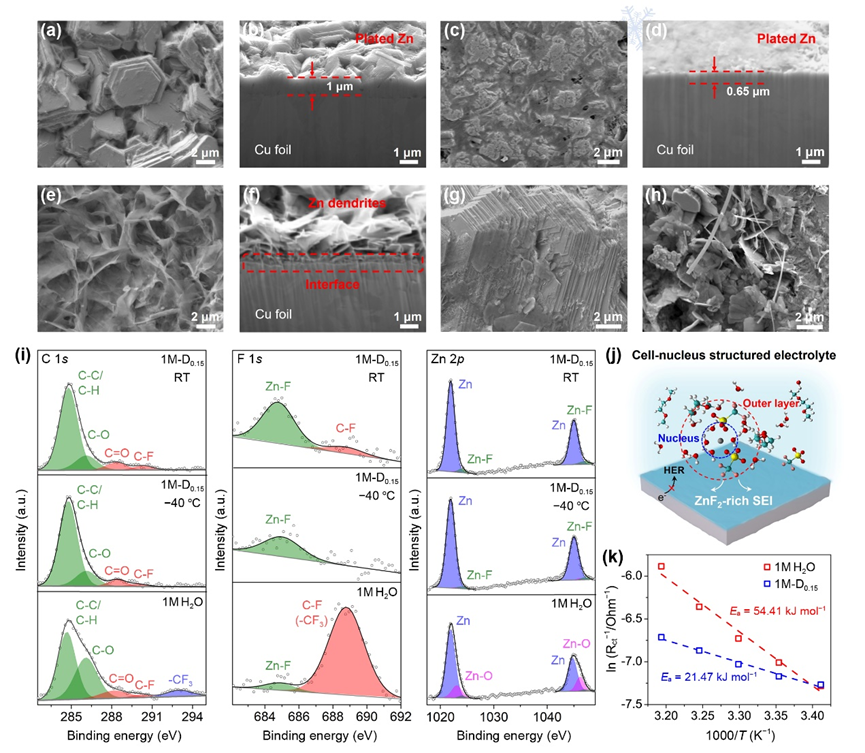
(a-f)在室溫和?40 oC低溫下,(a-d)1M-D0.15和(e,f) 1M H2O電解液中Cu基底上鋅沉積的FIB-SEM圖像;在(g) 1M-D0.15和(h)1M H2O中循環后鋅負極的SEM圖像;(i) RT和?40 ℃下Zn表面的C 1s、F 1s和Zn 2p信號XPS譜圖;(j)細胞核結構電解液中Zn負極界面化學示意圖;(k) 1M-D0.15和1M H2O中的去溶劑化能(Ea)對比。
▲利用FIB-SEM(聚焦離子束掃描電鏡)分析了不同體系中的鋅沉積形貌。1M-D0.15在室溫下的鋅沉積為六邊形的外延平行板塊狀形貌,由側視圖可看出沉積的鋅緊密附著在集流體表面,在?40 ℃低溫下也觀察到無枝晶和致密的鋅沉積。相比之下,1M H2O中顯示出明顯的枝晶生長和松散Zn沉積。對比Zn||Zn電池中10個循環后的鋅負極,最優體系同樣呈現平整的外延平行沉積,而純水體系中則出現明顯的枝晶生長與腐蝕等嚴重副反應。室溫和?40 ℃低溫下鋅負極表面的XPS證明富含ZnF2的SEI保護層的形成,大大降低了Zn2+的去溶劑化能壘。
圖5.低溫鋅全電池性能.
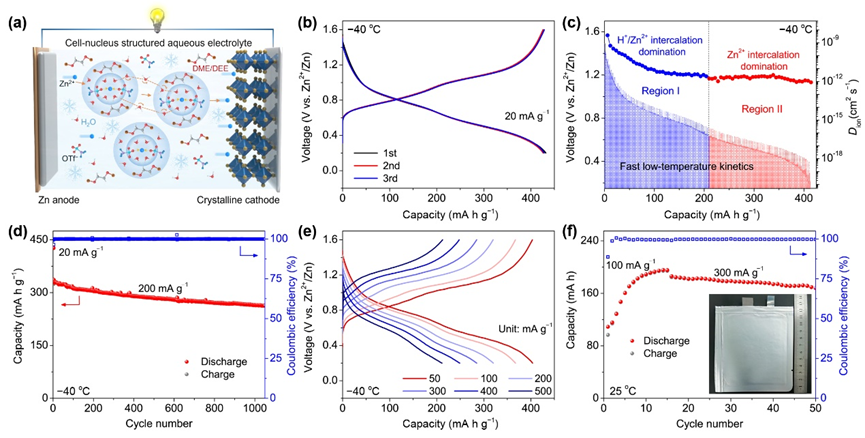
(a)?40℃低溫下鋅離子全電池工作示意圖。(b) 低溫下Zn|1M-D0.15|V2O5電池的充放電曲線;(c) 低溫下GITT放電曲線和相應的離子擴散系數;(d)低溫Zn||V2O5電池的長期循環穩定性和(e)倍率能力;(f) 室溫下Zn||V2O5軟包電池(插圖)的循環性能。
▲區別于目前報道的鋅-有機電池體系,我們將設計的細胞核電解液與傳統的V2O5正極組裝了全電池,測試其低溫下的性能。通過恒電流間歇滴定技術(GITT)測得最優體系即使在?40℃低溫下也能夠實現高擴散動力學,具有優異得低溫倍率性能(?40 ℃下,500 mA g?1提供212.4 mA h g?1容量),并能夠支持?40 ℃低溫下Zn||V2O5電池1000次以上的超長循環。為進一步評估電解液的實用價值,用其組裝了0.2 Ah級軟包電池,控制N/P比為3.56,可提供96.2 Wh kg?1能量密度(基于活性材料),室溫條件在0.3 A g?1下循環50次以上容量保持率高達91%。
研究總結
綜上,我們通過將低極性DME/DEE分子引入1 M Zn(OTf)2水溶液中,開發了具有富含OTf?的Zn2+溶劑化鞘內核(Zn2+-PSS)和DME/DEE調控的外溶劑化鞘層的“細胞核”結構耐低溫水系電解液。發現適當添加劑量的DME(即1M-D0.15)電解液表現出良好的低溫反應動力學,在?40 oC下離子電導率高達1.06 mS cm?1。DME的存在打破了水中原有的氫鍵網絡,將電解液的低溫凝固點降低至?52.4 oC。此外,富含OTf?的Zn2+-PSS不僅降低了水活度,還在鋅負極表面形成了富含ZnF2的SEI保護層,有效抑制了鋅枝晶的生長和副反應的發生。鋅負極在?40 oC低溫下表現出優異的可逆性和循環穩定性,在3 mA h cm–2的高面容量下(DOD = 51.3%)實現了3500 h的超長低溫循環穩定性,并能夠兼容V2O5/MnO2等傳統鋅電池無機正極材料,顯示出很好的應用前景。
審核編輯:劉清
-
鋅電池
+關注
關注
0文章
36瀏覽量
7796 -
電解液
+關注
關注
10文章
839瀏覽量
23063 -
DSC
+關注
關注
3文章
275瀏覽量
33544 -
DME
+關注
關注
0文章
22瀏覽量
7403 -
sei膜
+關注
關注
0文章
23瀏覽量
3535
原文標題:『水系鋅電』南開程方益教授&河大張寧教授JEC:“細胞核”結構電解液助力低溫水系鋅電池
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
武漢理工大學在水系鋅離子電池研究方面取得新進展
鎳氫電池的電解液是什么
新宙邦擬在美國投建10萬噸/年電解液項目
低濃度水系-非質子電解液助力鋅離子混合超級電容器設計


無機鋅鹽中非質子性極性溶劑適用原則的深入分析!

鋰離子電池電解液有什么作用?
揭示谷氨酰胺添加劑對高可逆鋅金屬陽極的多功能調節作用


調節用于高性能水系鋅離子電池的多金屬離子溶劑化結構





 工商網監
工商網監
評論