 莫一非團隊揭示固態電池中鋰金屬結晶機理
莫一非團隊揭示固態電池中鋰金屬結晶機理
【研究背景】
結晶是材料科學、物理和化學中的一個重要現象。當前人們主要研究由溫度或溶液變化引起的結晶過程,電化學沉積下的結晶過程仍然很少被研究,特別是下一代高能可充電電池中Li、Na、Mg和Zn金屬負極的電化學沉積和結晶過程依然鮮為人知。
在電化學沉積過程中,電解質中的金屬離子沉積并結晶為金屬晶體。過高的結晶能壘會提高電化學沉積的過電位。高過的電位或極化會降低電池性能和效率,甚至導致鋰枝晶的生成。通過降低過電位可以提高金屬負極的電化學性能。使用固態電解質來解決目前困擾金屬負極的問題是一個很有前途的方向。
固態電解質中金屬負極的電化學沉積行為跟與液體電解質中完全不同。在液體電解質中,電化學沉積過程中金屬顆粒的形核生長過程可以用經典的成核理論來描述。相比之下,在固態電解質上的鋰連續沉積過程中,這些沉積的鋰離子如何成為鋰金屬結晶的路徑和機理仍然不清楚。這種結晶過程具有固有的勢壘,并且會限制的電化學金屬沉積過程的速率。進一步改進金屬負極(如鋰金屬負極)的性能,急需從原子尺度上揭示結晶的原子路徑和動力學勢壘。
【工作簡介】
近日,美國馬里蘭大學莫一非教授團隊采用大規模分子動力學模擬方法,研究并揭示了固體界面處鋰結晶的原子路徑和能壘。研究發現,鋰結晶采用由界面鋰原子介導的多步路徑,具有無序和隨機密堆積構型作為結晶的中間步驟,這產生了結晶的能壘。這種對多步結晶路徑的理解將奧斯特瓦爾德分步規則(Ostwald's step rule)的適用性擴展到界面原子態,并提出了將有利的界面原子態作為中間步驟來降低結晶勢壘的合理策略。本文作者的研究結果開辟了界面工程的全新研究途徑,這將促進固態電池金屬電極的電化學沉積和結晶過程。該文章以“Lithium crystallization at solid interfaces”為題在線發表在國際頂級期刊Nature Communications上。現任同濟大學材料科學與工程學院特聘研究員楊孟昊是本文第一作者。
【內容表述】
為了研究固態電解質界面上金屬負極的電化學沉積行為。原子尺度建模方法在揭示固態電解質界面上原子轉變機制方面具有獨特的優勢,可以表征每個原子的能量并達到飛秒級別的實時分辨率。在本項研究中,本文作者使用固態電解質界面處的鋰金屬負極作為系統模型,采用大規模分子動力學模擬(MD)方法,以直接揭示固態電解質界面上鋰連續沉積過程中結晶的原子路徑和動力學勢壘。
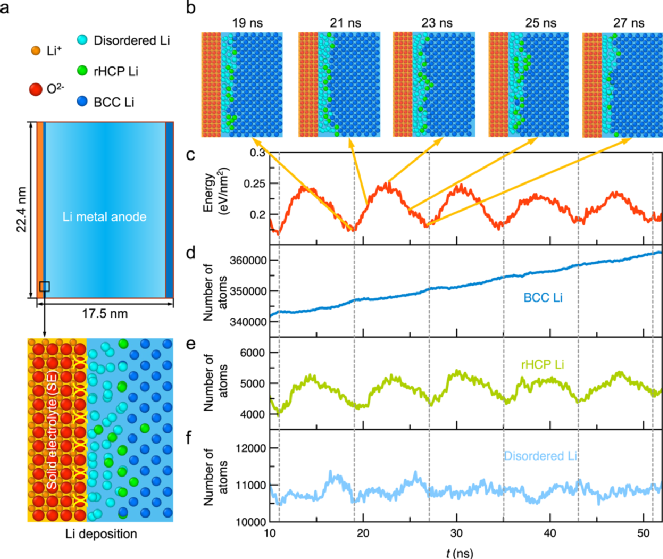
圖1. 鋰沉積過程中固態電解質界面鋰結晶的原子模型。a)分子動力學模擬中包含鋰金屬層(藍色)和固態電解質層(橙色)的原子模型;b)鋰沉積過程中一個能量變化周期下的Li-SE界面的原子結構;在鋰沉積過程中,c)以體相鋰金屬為參考時鋰金屬層的能量;d-f)鋰金屬層中不同局域構型下鋰原子的數量(如:體心立方(BCC)、隨機密排六方相(rHCP))。
Li-SE界面上界面原子結構在鋰電池循環過程中對金屬鋰的結晶過程中起著關鍵作用。通過采用分子動力學模擬方法跟蹤鋰隨時間的演變過程,作者進一步揭示了鋰從鋰離子到體心立方BCC-Li的逐步結晶過程。沉積的鋰原子首先被Li-SE界面上非晶鋰原子層吸附,隨后在后續的鋰沉積過程中通過兩條途徑結晶成BCC鋰金屬過程。
在第一種途徑中,沉積的Li原子首先通過disordered-Li,隨后轉變為BCC鋰金屬;在第二種途徑中,沉積的鋰原子通過disordered-Li之后,大部分的disordered-Li會形成下一個rHCP-Li中間相,最后轉變成為BCC-Li金屬(圖2b和d)。因此,鋰結晶由固態電解質界面處非晶原子層介導,其中界面原子、disordered-Li和rHCP-Li充當多步驟結晶過程的中間結構,而這些界面原子結構是由于固態電解質和鋰金屬之間界面相互作用引起的。
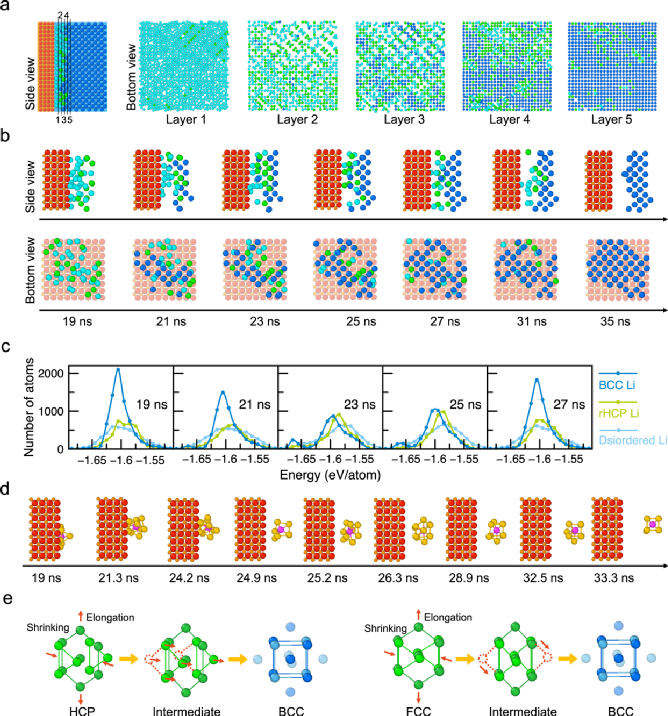
圖2. 鋰結晶的多步路徑。a) 19 ns時Li-SE界面的原子結構,以及展示的每層原子(無序disordered、隨機密排六方相rHCP、體心立方相BCC鋰分別以青色、綠色、藍色表示);b)一群鋰原子的結晶過程;d) 單個鋰原子(紫色)與它的近鄰原子們(黃色);c)鋰原子態密度(DOAS)展示了不同類型鋰的原子能量分布;e)密排六方相HCP或面心立方相FCC構型向體心立方相BCC構型演變的示意圖。
隨機密排六方相rHCP-Li構型 (HCP 或 FCC 的混合物) 通過小的鋰原子運動轉變為 BCC-Li,如圖2e所示。當HCP-Li轉變為BCC-Li時,{0001}六角面通過向<110>方向收縮或向<001>方向伸長變為{110}面,其他原子平行于六角面沿 <110> 方向移動形成 BCC構型。
FCC-Li以類似的方式通過小的鋰原子運動轉變為BCC-Li(圖2e)。除了具有較低的能量外,從rHCP-Li到BCC-Li的輕松轉變也使rHCP-Li成為鋰結晶過程中有利的中間態。這種原子路徑遵循奧斯特瓦爾德分步規則,即在最終穩定狀態之前形成更高能量但動力學上有利的中間態(圖3)。
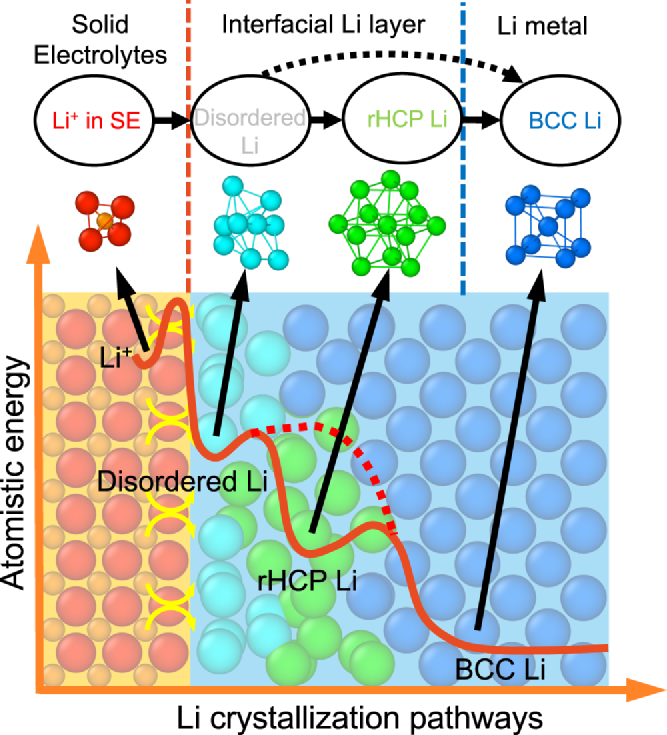
圖3. 鋰結晶的多步路徑示意圖。固態電解質中的Li+(桔色)先轉變為界面層中disordered-Li(青色)以及rHCP-Li(綠色),最后轉變為BCC-Li(藍色)。
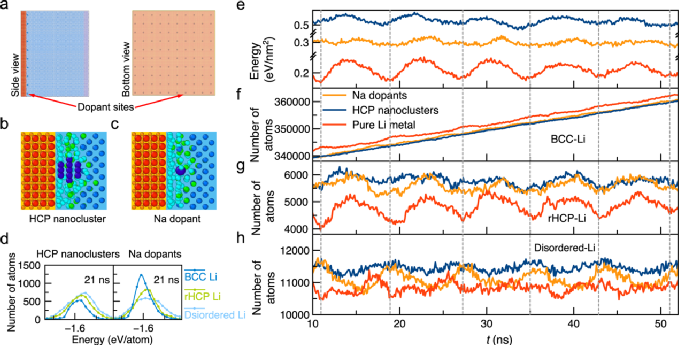
圖4. Li-SE界面處的鋰結晶。a)具有納米團簇和摻雜劑的Li-SE界面模型;具有b)HCP-Li納米團簇和c)鈉摻雜劑(深藍)的Li-SE界面的原子結構;d)不同結構類型鋰的原子能;e)具有納米團簇、摻雜劑和鋰金屬體相的鋰能量變化;f-h)鋰金屬中不同近鄰結構構型下鋰原子數目;原始的Li-SE界面(紅色)、有鈉摻雜時的界面(橘色)、有HCP-Li納米團簇時的界面(藍色)。
【核心結論】
分子動力學模擬結果揭示了固態電解質界面處鋰結晶的多步驟原子路徑,這表明奧斯特瓦爾德分步規則已經擴展到單個原子態。奧斯特瓦爾德分步規則表明:在結晶過程中,在熱力學穩定相之前首先形成較高能量的中間相。在鋰結晶的多步原子路徑中(圖3),高能界面原子態(例如disordered-Li和/或rHCP-Li)首先作為中間體形成,遵循奧斯特瓦爾德分步規則,然后轉變為體相晶體原子(即BCC-Li)。在這個復雜的多步結晶過程中,這些界面原子態的動力學和能量學可以通過界面原子的原子態密度(DOAS)來闡明。
作為結晶路徑中間體的界面原子態是鋰金屬和固態電解質之間界面相互作用引起的,因此可以通過界面工程進行調整。相比之下,在液體電解質中,結晶是由形核粒子和表面原子介導的,例如表面上的吸附原子或空位,如 Terrace-Ledge-Kink模型所示。這種從界面原子態的角度對多步結晶路徑的理解可以提出通過固態電解質的界面工程促進結晶的合理策略。這些調整結晶原子路徑的界面工程策略為提高固態金屬電池金屬負極的電化學沉積性能提供了機理解釋和理論指導。
審核編輯:劉清
-
電解質
+關注
關注
6文章
805瀏覽量
20018 -
HCP
+關注
關注
0文章
6瀏覽量
6879 -
固態電池
+關注
關注
9文章
692瀏覽量
27698 -
固態電解質
+關注
關注
0文章
83瀏覽量
5414
原文標題:馬里蘭大學Nat. Commun.: 莫一非團隊揭示固態電池中鋰金屬結晶機理
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
欣界能源發布全球首創480Wh/kg高能量鋰金屬固態電池
欣界能源發布“獵鷹”鋰金屬固態電池
固態電池中復合鋰陽極上固體電解質界面的調控

高能數造鋰金屬全固態電池小試級整線正式交付
固態鋰金屬電池的外部壓力研究

太藍新能源在固態鋰金屬電池領域取得技術突破



闡明鋰金屬電池中與溫度相關的鋰沉積/剝離過程以及非活性鋰的演變






 工商網監
工商網監
評論