 卡拉膠作為5V高壓LiNi0.5Mn1.5O4正極犧牲粘合劑!
卡拉膠作為5V高壓LiNi0.5Mn1.5O4正極犧牲粘合劑!
研究背景
尖晶石LiNi0.5Mn1.5O4(LNMO)正極材料具有較高的氧化還原電位和高能量密度(~650Whkg?1)。然而,LNMO的高工作電壓導致其與電解質發生副反應,導致正極界面的退化,導致循環時容量下降。碳酸鹽電解質在4.4 V 電壓下在正極表面形成厚且不穩定的正極-電解質間相(CEI),較厚的CEI層具有較高的界面電阻,從而損害了正極的循環穩定性。因此,LMNO正極在高電壓下的循環穩定性一直被認為是一種不可避免的問題。另一種有害的降解機制涉及六氟磷酸鋰(LiPF6),在高電壓下,LiPF6可以與微量的水反應生成氟化氫(HF),從而引發過渡金屬(TM)離子從正極表面的溶解和遷移。當裸露的LNMO表面暴露在電解質中時,TM離子的溶解會加速。溶解的TM離子沉淀在負極/正極,以金屬粒子形式的表面進一步催化兩個界面上副反應。迄今為止,已經提出了各種抑制電解質副反應的策略,如使用電解質添加劑、自由基清除劑、和界面保護層。然而,完全覆蓋活性粒子是不可行的,即使可以實現完全覆蓋,也會提高界面阻力。自由基清除劑不能完全消除HF分子。另一方面,粘結劑具有保護活性電極層和集流體之間的粘附力,因此,我們假設粘合劑與電解液可能對正極界面的形成和性能產生重大影響,而正極界面與電池的關鍵電化學性能指標密切相關。
雖然聚(偏氟乙烯)(PVDF)是最廣泛使用的粘合劑層狀金屬氧化物正極,但并不適用于LNMO正極,因為其不能均勻覆蓋活性物質,可能無法解決上述界面問題。作為PVDF的替代品,生物聚合物和合成聚合物利用它們的羥基或負電荷官能團分別參與與LNMO表面的氫鍵和離子偶極相互作用。這些相互作用排列均勻地覆蓋LNMO表面,從而防止副反應發生。然而,這些粘合劑的均勻覆蓋,反過來又增加了界面電阻,因為從結構的角度來看,它們對鋰離子的電導率造成不利影響。粘合劑有一個關鍵的功能是影響鋰離子擴散到CEI層,氧化弱官能團粘結劑在高壓下可以轉換成某些部分CEI層。考慮到這一原理,可以選擇粘合劑的官能團,使分解后剩余的部分形成CEI配合物/粘合物,以促進鋰離子在CEI中的擴散。因此,粘合劑可以表現為“犧牲性的”。雖然電解質添加劑也可以設計出類似的目的,但如果其最低未被占用的分子軌道水平低于負極的工作電位,則干擾負極界面。顯然,粘合劑的主要作用必須通過利用其他官能團來保持其粘附性能,強調多功能性作為LNMO正極粘合劑的關鍵設計原則。
成果簡介
近日,首爾國立大學/韓國能源研究所等合作者為了解決LNMO高電壓循環下容易發生界面降解副反應,從而阻礙長期和高倍率循環穩定性等問題。在此,作者通過摻入犧牲性粘合劑,即 λ-卡拉膠 (CRN)(一種硫酸化多糖),克服了LNMO長期循環性較差的問題。這種粘合劑不僅通過氫鍵和離子偶極相互作用均勻地覆蓋LNMO表面,而且提供了含有LiSOxF的離子導電正極-電解質界面層。利用這兩個特性,CRN基電極的循環性能和倍率性能遠遠優于基于傳統的聚(偏二氟乙烯)和海藻酸鈉粘合物。本研究引入了一種新的概念,即“犧牲”粘合劑,用于鋰離子電池電極制造,使其具有優越的電化學性能。該工作以“Carrageenan as a sacrificial binder for 5 V LiNi0.5Mn1.5O4 cathodes in lithium-ion batteries”為題發表在Advanced Materials上。
研究亮點
(1) 通過使用硫酸鹽多糖粘合劑,即CRN,克服了LNMO電池在高壓循環條件下穩定性較差的問題;
(2) 粘合劑形成CEI/粘合物,通過高壓條件下硫酸鹽基的不可逆氧化分解對鋰離子具有高導電性;
(3) CRN的親水性使其可行的覆蓋LNMO粒子均勻,但硫酸基的氧化分解導致CEI/粘合劑復雜層包含LiSOxF,促進鋰離子傳導;
(4) 本研究是首次在粘合劑設計中演示“電化學犧牲”概念來控制CEI層。
圖文導讀
CRN在其主主鏈上同時有硫酸基和羥基(圖1a),使其能夠同時作為犧牲劑和粘合劑。這兩種功能在5V 高壓下同時解決LNMO正極兩個挑戰性問題:基于正極界面的強粘附性和高鋰離子傳導,具有良好的活性粒子覆蓋范圍。為了證明CRN這兩個官能團的作用,我們選擇了海藻酸鈉(ALG,圖1b)和PVDF(圖1c)作為對照結合劑。ALG在結構上與CRN高度相似,只是ALG沒有硫酸鹽基團。ALG長期以來一直被用作正極和負極的水性粘結劑。利用透射電子顯微鏡(TEM)研究了原始狀態下LNMO表面粘合劑的覆蓋情況,CRN和ALG粘結劑沿LNMO表面的覆蓋范圍較薄且均勻(圖1d,e),這歸因于它們的親水官能團。相比之下,PVDF的分布并不均勻,因為它在LNMO表面局部聚集(圖1f)。這是因為PVDF具有弱范德華相互作用,沒有引起與活性材料的粘附。
為了證明CRN中的硫酸鹽官能團確實被電化學分解,在第一個和第三個形成周期中,在半電池中進行了電化學浮充試驗(圖1g-j)。在這些浮充測試中,在每個電壓水平上施加恒定電位10小時,同時監測特定的電流。第一個形成周期的充電過程的特征是ALG基和PVDF基電極都有連續的無峰曲線(圖1g)。然而,對于基于CRN的電極,在4.8 V時觀察到一個肩峰(圖1g中的箭頭),這在圖1h的放大圖中更明顯。僅在CRN基電極的曲線上出現這個肩峰是由于其硫酸鹽官能團的氧化分解,而不是與電解質分解相關。在第三個形成循環中,所有電極的電流曲線形狀相似(圖1i,j),這表明在初始充電過程中CRN的氧化是不可逆的,之后電極穩定。這一結果與電解質添加劑的報道一致;從CRN中提取的硫酸衍生物在初始充電過程中在LNMO表面形成,一旦在早期循環中建立了電子絕緣界面層,就不會被進一步氧化。這些一系列的結果表明,CEI/粘合劑配合物的穩定性在第一次循環中形成,粘合劑在該配合物的穩定性中起著至關重要的作用。
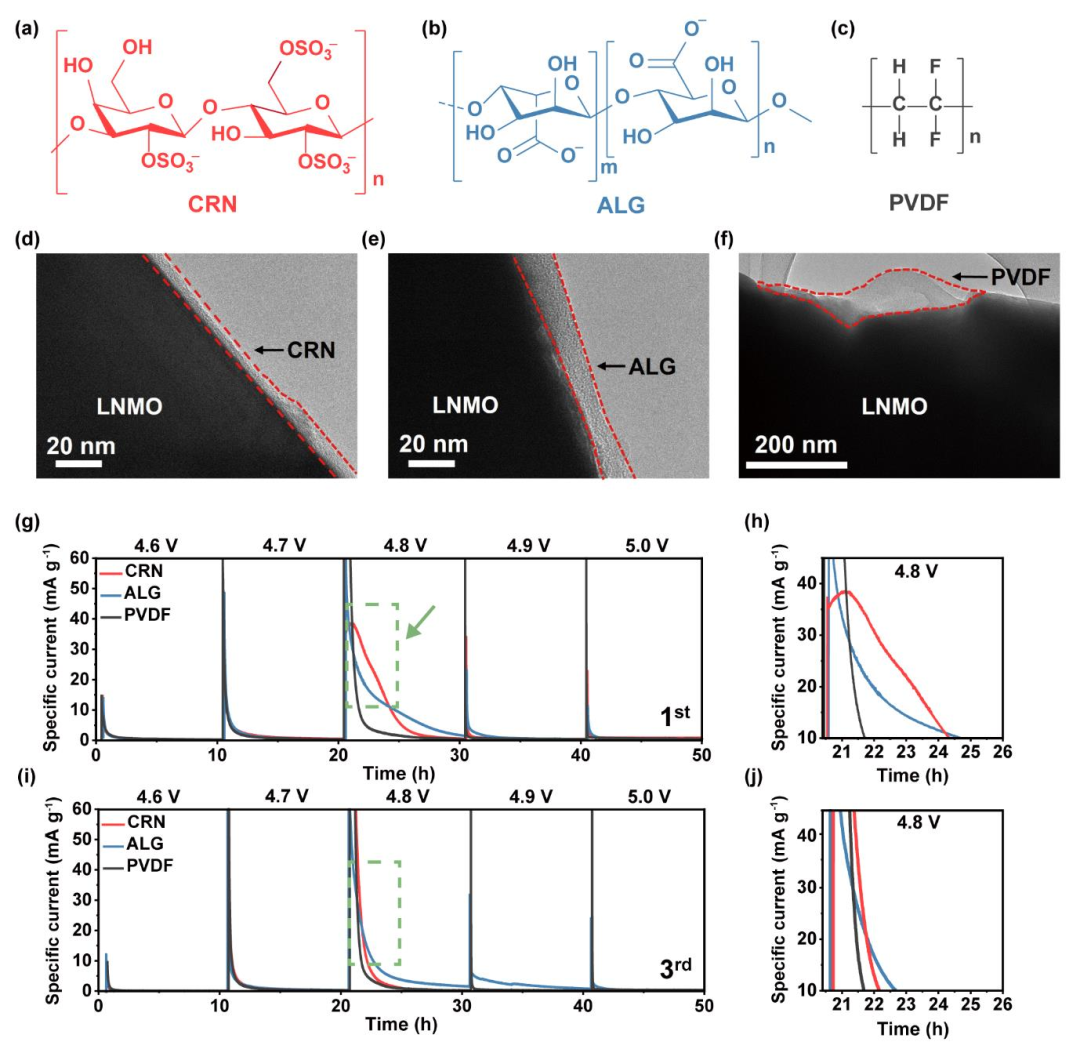
圖1.CRN、ALG和PVDF聚合物粘合劑的理化和電化學性質。(a) CRN、(b) ALG和(c) PVDF的化學結構。在原始狀態下,LNMO表面上的(d) CRN、(e) ALG和(f) PVDF的透射電鏡圖像。紅色虛線表示粘合劑所覆蓋的表面區域。在(g和h)第一次和(i和j)第三次充電時,CRN、ALG和PVDF基電極的電化學浮動試驗周期(h和j)分別由(g和i)中綠色虛線劃分的區域的擴大
圖2a顯示了三組電池在經過活化3圈后在1C(1C = 147 mA g?1)和室溫下在3.5-5.0V電位范圍下的循環性能。基于CRN的電池在循環性和比容量方面都比基于ALG和PVDF的電池表現出優越的性能。基于CRN-、ALG-和PVDF-的電池分別提供了110.7、96.1和112.0mAhg?1,在900次循環后保留了100.0%、98.6%和75.4%的原始容量。三組電池的平均CEs分別為99.6%、99.6%和99.4%。多糖粘合劑對LNMO表面的均勻覆蓋可以緩解表面降解,從而更有效地維持循環。硫酸基分解引起的CEI的離子導電特性使其具有更高的比容量。粘合劑也會顯著影響倍率性能。在0.1C到5C之間,基于CRN電池的放電容量保持得要高得多(圖2b),明顯利用了其CEI/粘合劑配合物高的鋰離子電導率。例如,在5C時,CRN、ALG和PVDF基細胞的比容量分別為81.3、59.7和36.3 mAh g?1。通過電化學阻抗譜(EIS)對原始狀態和100個循環狀態的分析,闡明了三組電池之間不同的倍率性能(圖2c,d)。CEI電阻(RCEI)和電荷轉移電阻(RCT)與LNMO表面的界面電阻密切相關。在原始狀態下(圖2c),CRN基電極比PVDF基電極表現出更高的界面電阻(CRN基和ALG基電極的界面電阻分別為305.0和343.1Ω),而PVDF基電極為139.5 Ω)。經過100個周期后(圖2d),基于CRN的細胞的RCEI和RCT(4.5和88.9 Ω)分別低于基于ALG電極(分別為7.2和155.1Ω)和基于PVDF的細胞(分別為64.2和252.9Ω)。這一結果再次反映了CRN中硫酸鹽基團形成的CEI/粘合劑配合物的鋰離子電導率較高。在100次循環中,PVDF電極的RCEI和RCT均顯著增加,這反映了PVDF覆蓋范圍有限導致界面不穩。此外,在0.05到1 mV-1的不同掃描速率下的CV被用來研究鋰離子在LNMO表面的傳輸。鋰離子擴散系數(DLi+)可以從ip和v1/2之間的關系中提取出來,用如下式表示:
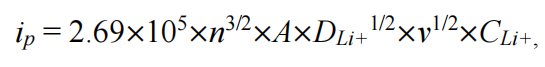
ip是峰值電流,n是電子反應的數量(n=1),A為電極的表面積,v是掃描速率,CLi+為鋰離子的體積濃度(0.02378 mol cm–3)。利用ip與v1/2圖的斜率,計算出基于CRN-、ALG-和PVDF-電極的DLi+值分別為2.84×10-10、1.81×10-10、8.98×10-11 cm2s-1(圖2e)。這些結果與倍率性能和EIS結果所表現出的趨勢一致,并歸因于鋰離子通過CRN基電極的鋰離子導電CEI/粘合物的高效遷移。
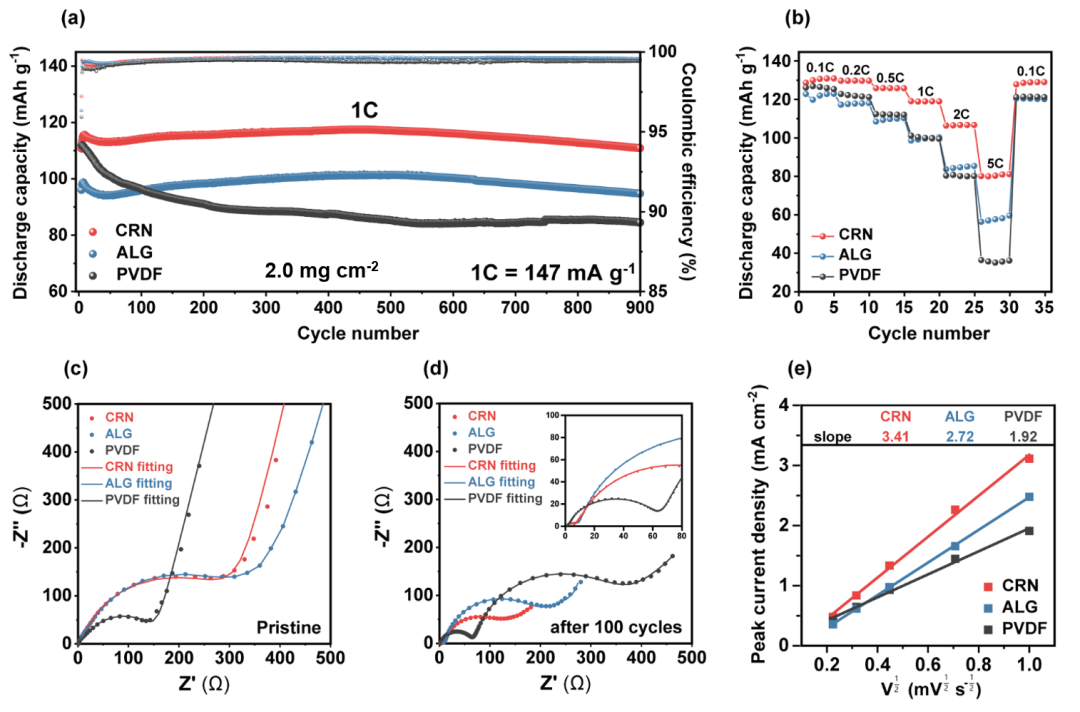
圖2. CRN-、ALG-和PVDF-基電極的電化學性能。(a)在3.5-5.0V(14.7 mA?1)下進行900個循環的放電容量和CEs。半電池0.1C(14.7 mA g?1)下形成三次循環。(b)在不同倍率下的倍率性能。奈奎斯特圖在原始狀態的(c)和100個循環后擬合。(e)峰值電流(ip)與CV數據中掃描速率(v1/2)的平方根之間的關系。
圖3a-c分別顯示了CRN-、ALG-和PVDF-基電極在三個不同循環階段的F 1s XPS譜:原始階段、三個形成循環后和100個循環后。在原始狀態下,CRN-和ALG-基電極沒有觀察到信號;然而,在PVDF基電極中觀察到一個屬于PVDF的強C-F鍵的峰值。經過三個形成周期后,在基于CRN-和ALG-的電極上觀察到與氟化鋰相關的峰(圖3a,b),這是由于PF6-陰離子的分解。CRN-電極在687.98 eV處出現了一個峰值,分配給LiSOxF。通過與商業LiSO3F的比較,證實了該峰的存在。相比之下,這些無機物的峰在PVDF基電極的光譜上沒有明顯的特征(圖3c)。經過100個循環后,CRN-和ALG-基電極中的氟化鋰下降(圖3a,b)。CRN-基電極中的LiSOxF也降低,相比之下,PVDF-基的電極在相同的循環期間,氟化鋰和LixPOyFz均表現出更大的增加(圖3c)。考慮到XPS的探測深度約為5nm,預計在循環的進行,CRN-和ALG-基電極早期形成的氟化鋰逐漸被其他CEI覆蓋。與此相反,在PVDF基電極的情況下,氟化鋰和LixPOyFz都隨著循環的增加而增加。
在其S 2p XPS結果中進一步捕獲了CRN基電極的CEI特性。在原始狀態下,觀察到與O-SO3鍵相關的雙峰峰(171.17和169.97eV)(圖3d),反映了裸CRN中的硫酸鹽基團。3個循環后,S 2p的LiSOxF峰值和O 1s XPS譜下降,類似于多糖電極的氟化鋰峰,表明這些組分在LNMO表面形成,但在以后的周期中幾乎沒有形成。此外,即使在循環后,CRN基電極中的硫酸鹽(O-SO3)峰仍然被檢測到,這意味著CRN中并非所有的硫酸鹽都被消耗掉以形成LiSOxF。二次離子質譜飛行時間(ToF-SIMS)也檢測到LiSOxF的存在(圖3e,f)。對離子碎片的質譜分析顯示,LiSO3F-(m/z=105.97)和LiSO2F-(m/z=89.98)以負離子形式存在,作為LiSOxF的特征。電化學浮動測試結果、O 1s、F 1s和S 2p XPS結果以及ToF-SIMS結果一致支持我們的觀點,即CRN電極中的CEI層包含高壓下CRN硫酸官能團分解的LiSOxF。
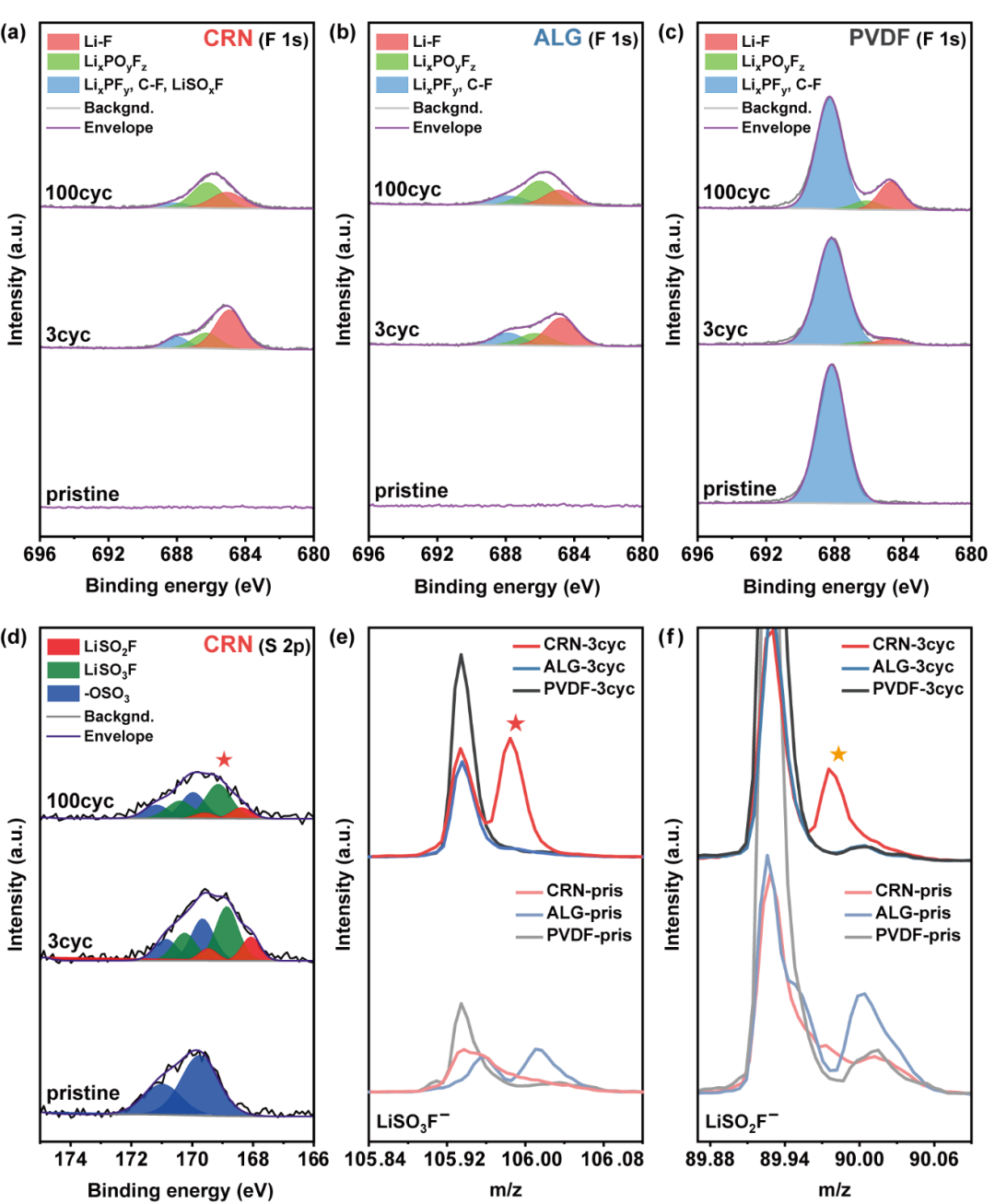
圖3.CRN-、ALG-和PVDF基電極中CEI組分的識別。本研究中(a) CRN-、(b) ALG-和(c)PVDF基電極的F 1s XPS結果在原始狀態,在0.1C下三次循環,在1C下100次循環。(d) S 2p XPS結果在原始狀態下,在0.1C下三次循環,在1C下100次循環。(e) LiSO3F?和(f) LiSO2F?二次離子片段在原始狀態下的ToF-SIMS質譜。星號標記的峰代表LiSO3F?(紅色)和LiSO2F?(橙色)離子片段(Li=7.016,S=31.972,O=15.994和F=18.998)的m/z特征值。
追蹤其他陰離子LiSO2F- 和LiF-(m/z=26.00)碎片,作為濺射時間的函數(圖4a)。與S 2p XPS結果一致,LiSO2F- 陰離子片段的強度隨著濺射開始開始增加,40秒后陰離子片段的強度逐漸下降,表明其更集中地存在于CEI內部。仔細觀察,在CRN基電極中,濺射40秒后LiSO2F-陰離子片段的強度高于濺射前(圖4b)。這與基于ALG-和PVDF的電極不同(圖4c,d),它們在相同的濺射時間內的峰值強度幾乎沒有差異。與CEI/粘合物的氟化鋰成分相關的LiF-陰離子片段的深度剖面也顯示了類似的異常。與基于CRN和ALG的電極不同,其強度在初始40秒內增加,然后逐漸下降。基于PVDF的電極中的LiF-的強度從一開始就單調下降。從三個電極在0和40秒時的鋰離子片段的強度曲線上可以明顯地看出這些趨勢(圖4e-g)。同樣,該陰離子片段的峰值強度與CEI/粘合物內部存在的濃縮氟化鋰有關。ToF-SIMS深度剖面數據描繪了一個關于CRN基電極的CEI層的一致圖像。也就是說,CEI層的特征是在該層的內部形成具有有益的LiF和LiSOxF,這在保護電極免受有害的副反應方面起著重要的作用。根據目前所得到的分析結果,三個電極的CEI層的組成和結構如圖5所示。
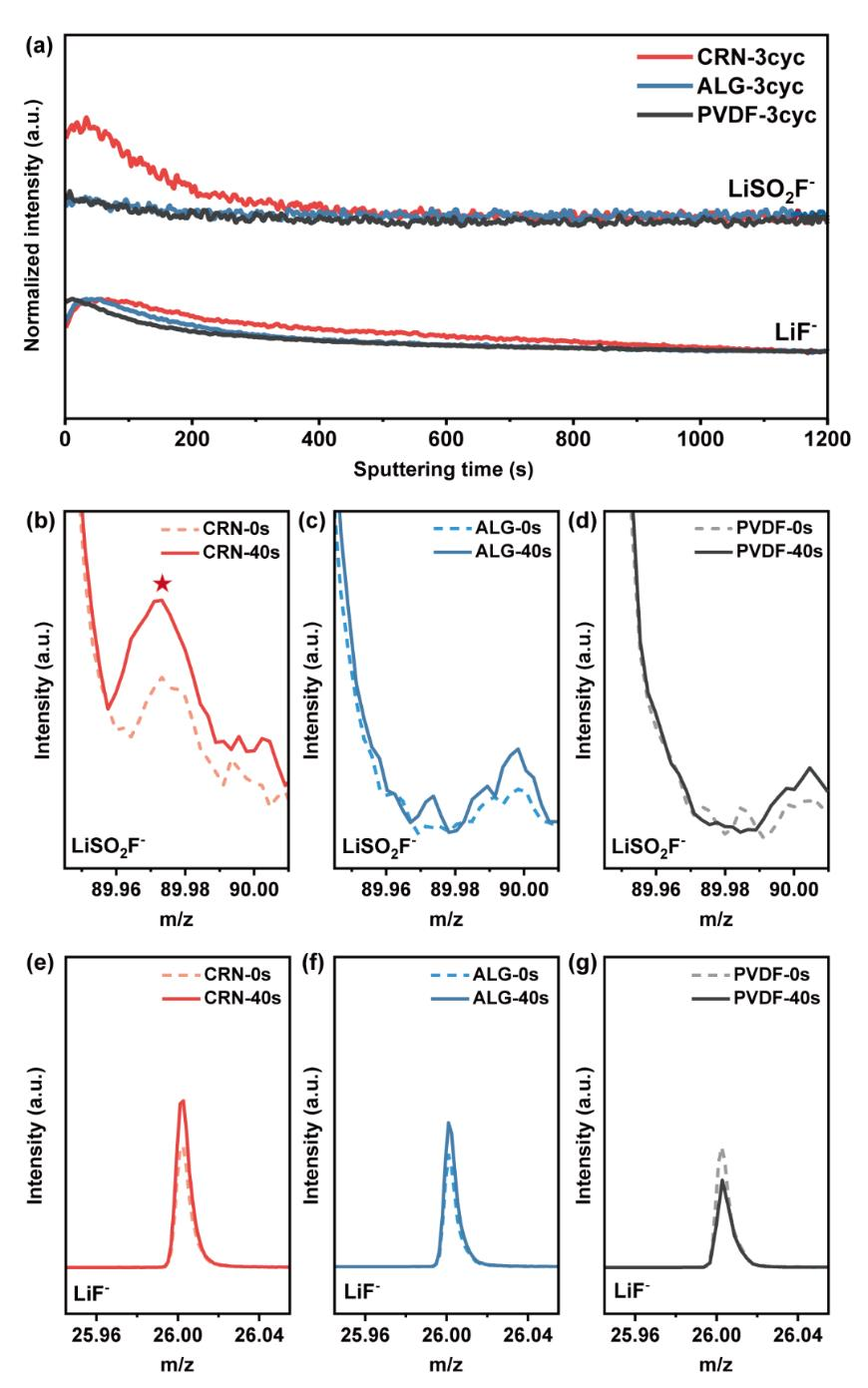
圖4. 無機CEI組分在CRN-、ALG-和PVDF-基電極中的不同位置。在0.1C下,三個粘結劑電極的LiSO2F?和LiF?二次離子片段的(a) ToF-SIMS深度分布。在0和40秒時,(b) CRN-、(c) ALG-和(d)PVDF電極中LiSO2F?二次離子片段的ToF-SIMS譜。(e) CRN-、(f) ALG-和(g)PVDF基電極中LiF?二次離子片段的ToF-SIMS譜。
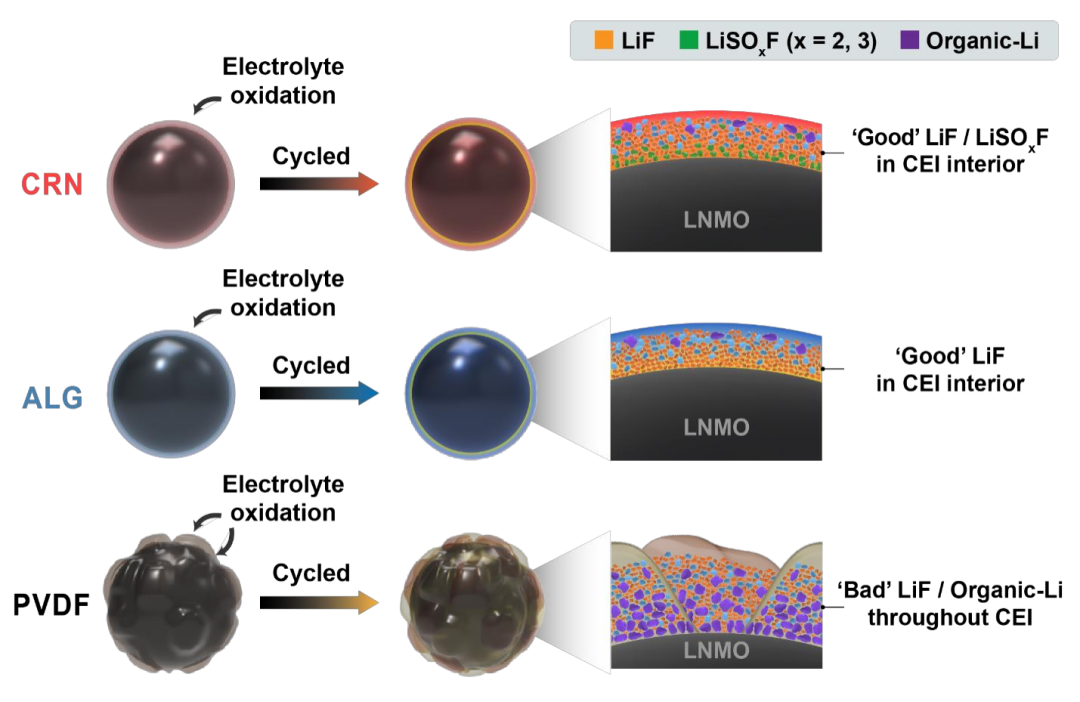
圖5.PVDF-、ALG-和CRN基電極的粘合劑覆蓋范圍和CEI形成的示意圖
在鋰離子擴散方面,CRN基電極CEI層內部的LiSOxF/LiF的組合組成比ALG基電極CEI層內部的LiF更有利。這與CRN基電極優越的放電容量和倍率性能相一致。這些比較結果使我們分別考慮LiSOxF和LiF的鋰離子和反陰離子SOxF?和F?之間的靜電學。因為靜電力會影響CEI層中鋰離子遷移率。為此,通過密度泛函理論(DFT)計算,計算了鋰離子與反陰離子的結合能(圖6a)。計算結果表明,CRN中的硫酸鋰衍生物(LiSO3F和LiSO2F)的結合能分別為-6.27和-6.59eV,均低于氟化鋰(-8.11eV),說明Li離子相互作用與LiSOxF的陰離子比LiF強。此外,利用固態7Li核磁共振波譜(7LiNMR)對商用LiSO3F和氟化鋰進行了分析,實驗驗證了上述計算結果(圖6b)。根據7Li-NMR分析,LiSO3F的化學位移(-1.44ppm)低于氟化鋰(-0.85ppm),說明LiSO3F中Li原子附近較高的電子密度進一步屏蔽了Li原子的核不受外場的影響。這一結果與上述DFT計算結果相關聯,即LiSO3F的陰離子與鋰離子的結合較弱,從而使鋰離子可以更自由地擴散。
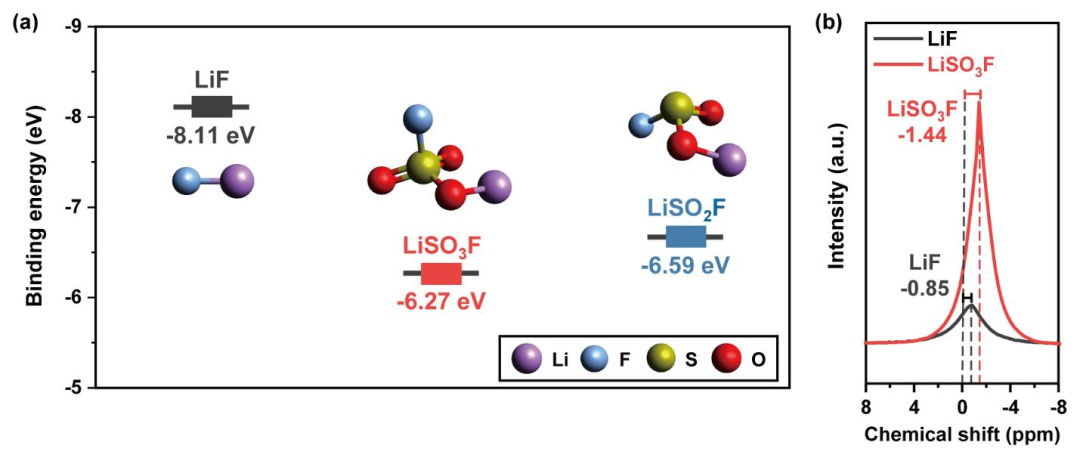
圖6. LiF和LiSOxF(x = 2,3)CEI組分的鋰離子結合親和力。利用GAUSSIAN16軟件包,通過DFT計算得到LiF和LiSOxF(x = 2,3)的(a) Li離子結合能。氟化鋰和LiSO3F的(b) 7Li MAS核磁共振譜。
總結與展望
在這篇文章中,作者通過使用硫酸鹽多糖粘合劑,即CRN,克服了LNMO正極在高電壓下表面降解導致循環穩定性較差的挑戰。這種粘合劑形成CEI/粘合物,通過高壓條件下硫酸鹽基的不可逆氧化分解對鋰離子具有高導電性。CRN的親水性使其均勻覆蓋LNMO粒子,硫酸鹽基的氧化分解導致CEI/粘合劑復雜層包含LiSOxF,促進鋰離子傳導,解決LNMO正極在高倍率和長循環條件下的問題。本研究是首次在粘合劑設計中演示“電化學犧牲”概念來控制CEI層。基于同樣的原理,可以考慮各種官能團來靶向改性鋰離子電池正極,解決界面不穩定性帶來的不利影響,使這些電極具有優越的電化學性能。
審核編輯:劉清
-
鋰離子電池
+關注
關注
85文章
3215瀏覽量
77550 -
電解質
+關注
關注
6文章
805瀏覽量
20017 -
XPS
+關注
關注
0文章
97瀏覽量
11968 -
DFT算法
+關注
關注
0文章
27瀏覽量
7524
原文標題:首爾國立大學/韓國能源研究所Advanced Materials:卡拉膠作為5V高壓LiNi0.5Mn1.5O4正極犧牲粘合劑!
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
探究全電池容量衰減的根本原因
LG化學進軍汽車用粘合劑市場
漢高華南應用中心兩周年!粘合劑助力手機屏占比、AI散熱探索,材料創新無極限
DELO推出可靠密封圖像傳感器的新型粘合劑
ISO 8510-2 粘合劑-膠粘劑180°剝離試驗機(軟質與硬質粘合試樣)

TYPC USB作為DEVICE使用的話,VBUS需要接5V電壓嗎?
5V Tolerant I/O的意思是STM32和5V供電的芯片可以直接通信嗎?
5V+10%輸入,1.5KV隔離 5V/2W 單路輸出解決方案VP8504B003數據手冊
漢高:碳化硅、HBM存儲等高成長,粘合劑技術如何助力先進封裝

圍壩膠需要多長時間才能固化?

DELO新型電子粘合劑促進自動駕駛技術發展
電子元件的“粘合劑”—激光錫焊介紹

一種有機-無機非對稱固態電解質,實現長循環穩定的高壓鋰電池

4.7V高穩定鋰離子電池用HBCHHI添加劑輔助商用酯電解液






 工商網監
工商網監
評論