 基因組重測序的應用領域有哪些
基因組重測序的應用領域有哪些
背景介紹
1.1
基因測序在癌癥領域上的應用
癌癥是目前人類所面臨的最大敵人,其發病率隨著年齡增長而升高,在人口老齡化加劇的當下,全社會的癌癥負擔也將愈發嚴峻。癌癥難以治愈的原因之一是腫瘤具有強異質性,即相同癥狀、相同病理改變卻可能由完全不同的基因變化而造成,以至于同類型癌癥患者對于相同藥物的藥效反應有很大的差別 。這些基因上的變化包含了單堿基突變(SNV)、小片段插入缺失(InDel)、結構與拷貝數變異(SV & CNV)和甲基化等。隨著近幾年基因測序成本如圖 1所示不斷下降,在萬元內即可完成人類的全基因組測序,GPU的技術發展也帶來分析成本與時間的下降,于是用于檢測基因組變化的重測序技術在癌癥治療中起到了越來越重要的作用。基因組重測序的應用主要有三個方向:
01
基因測序技術提供腫瘤內基因分子水平的變化,為研究提供預見性,支持腫瘤新靶向藥物研發。
02
提高腫瘤早期診斷準確率,降低高危風險,從而提高癌癥存活率。以在兒童中高發的兒童神經母細胞瘤(NB)為例,目前國內低、中危組NB患兒,長期無瘤生存率可達90%,但高危組NB患兒的5年總生存率不足30%。而如果能夠做到早期診斷的話就可以通過手術切除聯合化療的方式進行治療,從而提高治愈率。
03
基于易感基因分子水平的基因變化檢測,可以根據變化情況為患者提供個體化和預見性的治療。
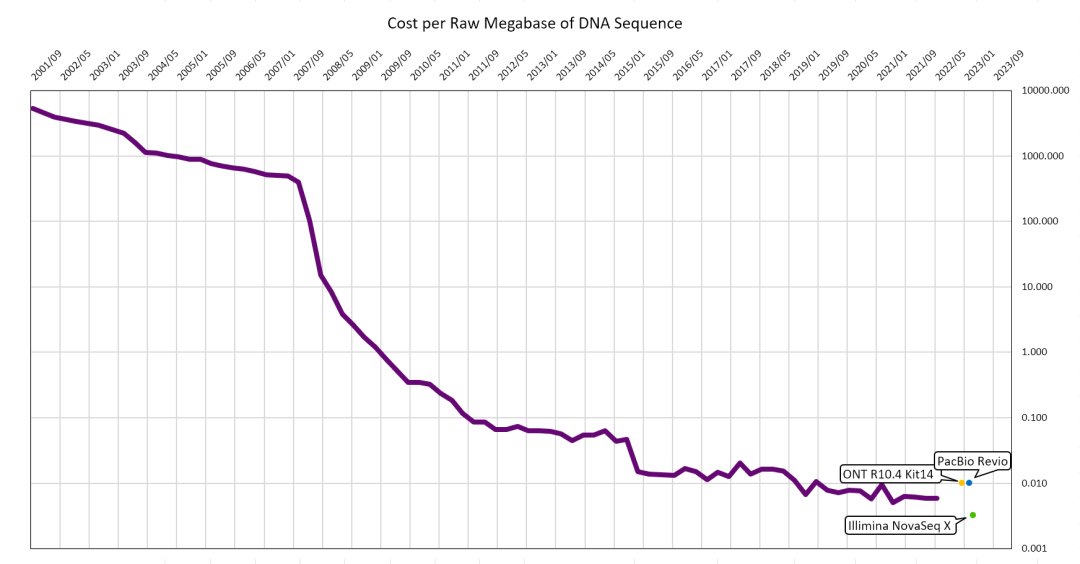
圖 1 每兆數據DNA測序開銷
圖中數據來源于Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) 和 Next-Generation-Sequencing.v1.10.25
1.2
基因測序手段介紹
當下基因組重測序常見的測序手段分為以Illumina雙端150bp為代表的短讀長測序與以PacBio HiFi或Oxford Nanopore Technology(ONT)為代表的長讀長測序兩種。如圖1所示,Illumina雙端測序將DNA打斷兩端加上不同接頭與流動池中分布的接頭結合不斷合成、退火與擴增,擴增后利用帶熒光的dNTP與流動池中的DNA鏈結合,由于不同的dNTP發出不同熒光,于是可以通過掃描流動池中的熒光信號,得到序列信息;PacBio 將單分子與DNA聚合酶固定在零模波導孔上,激光從孔的底部照入,激光不能穿過小孔且只能照亮孔底的一小部分區域。當DNA聚合酶使用游離的dNTP進行聚合反應時,dNTP上的熒光基團被激光激發,通過傳感器將光信號轉化為電信號。ONT則是通過讓 DNA 單分子通過帶電的納米孔,不同的堿基使得電流發生改變,傳感器記錄電流變化,測序儀將電流變化轉化為堿基值。
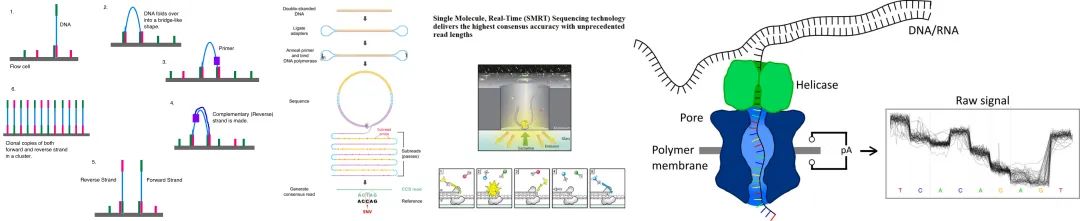
圖 2 測序原理示意圖 (a) Illumina 雙端測序,
圖片來源于DMLapato, Illumina dye sequencing, https://en.wikipedia.org/w/index.php?title=Illumina_dye_sequencing&oldid=1159305769#cite_note-Illumina,_Inc_2013-7
(b) PacBio 零模波導孔單分子實時測序,
圖片來源于百邁客生物, pacbio三代測序儀, http://www.biomarker.com.cn/technology-services/pacbio
(c) ONT 納米孔測序,
圖片來源于 Kraft, Long-read sequencing in human genetics, DOI: 10.1007/s11825-019-0249-z
短讀長測序的優勢在于價格低廉與測序堿基準確度高,長讀長測序擁有以下幾個優點:
01
長讀長測序使用單分子測序,不需要擴增反應。一方面節省了時間,另一方面也避免了擴增中引入的錯誤,比如擴增效率不同帶來的偏好性問題。
02
長讀長測序相比于短讀長測序更容易跨過基因組的重復區域。在短讀長測序結果中,至少748個與人類相關的基因存在測序暗黑區域,這個區域序列的Read會出現低深度、低覆蓋度及低比對質量。而造成這個現象的主要原因是在參考基因組中該區域的序列多次重復。而測序序列長到能夠跨過重復區域的長讀長測序正好解決了這個問題。
03
長讀長測序相比于短序列可以更容易得到核酸的甲基化水平。短讀長測序基于熒光信號,通常需要將樣本分為兩份,一份利用亞硫酸氫鹽處理將未甲基化的C轉化為U,另一份直接測序進行比較獲得基因組甲基化位點。而基于電信號的三代測序可以直接區分出甲基化的C與未甲基化的C,避免了亞硫酸氫鹽等藥品處理和多次測序引入的錯誤。
而近幾年來測序的技術有兩個明顯的發展趨勢:
01
短讀長測序和長讀長測序成本均逐步降低。短讀長測序成本降低幅度變緩,長讀長測序的價格隨著儀器的升級已經降到了三年前短讀長測序成本。在一些應用場景中,比如結構變異的檢測,由于長讀長測序只需要三分之一短讀長測序的數據量,長讀長測序的成本已經低于短讀長測序。
02
隨著試劑與算法的研發,長讀長測序的準確度也逐漸接近短讀長測序。在單堿基質量和測序價格的保證下,長序列能保留更原始的基因組特征。
于是目前研究所選擇的測序手段逐漸從短讀長測序向長讀長測序轉移。
1.3
基因組重測序的流程介紹
如圖3所示,重測序的流程主要包含以下大步驟:
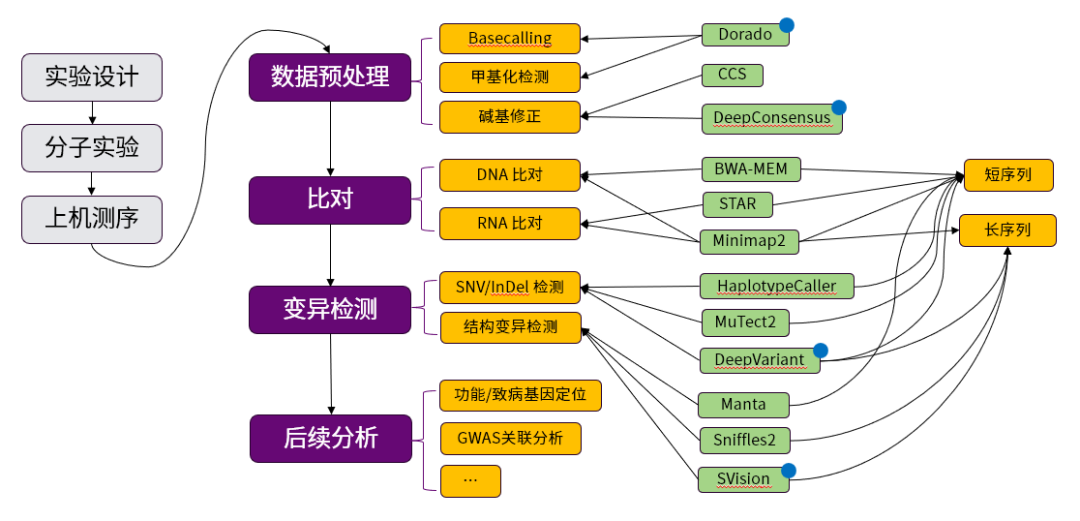
圖 3 基因組重測序流程與軟件示意圖。
圖中紫色矩形部分為主要分析步驟,黃色矩形部分為分析類別,綠色矩形部分為軟件,藍色上標的軟件為利用到了機器學習的軟件。
01
實驗設計、分子實驗與上機測序:這一步包括了需求分析和選擇測序手段。分子實驗通常包括核酸提取、核酸分子打斷和樣品的制備。上機測序將樣品加入測序儀中,測序儀產生序列信息,完成測序。
02
數據預處理:通常測序儀會利用傳感器捕獲電信號,測序儀的Basecalling步驟和甲基化檢測步驟將電信號轉化為文本形式的序列。基因組測序的結果保存檢測得到每一個核酸分子的堿基和該堿基的質量值,ONT測序和PacBio測序也會保存原始電信號。甲基化檢測的結果會同時輸出每一個堿基的甲基化概率。長序列檢測會發生隨機的測序錯誤,被認為準確度不高,需要進行堿基修正,修正測序錯誤來提升堿基質量值。
03
比對:這一步驟將測序得到的序列與參考基因組比對,確定該序列來自于參考基因組的哪個部分。RNA測序得到的序列相比于DNA缺失了內含子部分,長讀長測序與短讀長測序在錯誤率、復雜度上有區別。所以軟件上會根據樣本的類型和測序序列長度做區分。
04
變異檢測:變異檢測將獲得樣本與參考基因組的差異,這些差異包括了單堿基的變異(SNV)、小的插入缺失(InDel)以及大的結構變異(SV)。短讀長測序由于單堿基的準確度高,在SNV和InDel上有優勢,而對于長度大于短讀長測序序列長度的結構變異而言,長讀長測序更有優勢。所以軟件上會根據需要檢測的變異長度和測序序列長度做區分。
05
后續分析:后續分析則包括了變異的過濾、注釋、比較以及關聯分析等過程,這一步將以萬計的變異中篩選出與研究課題息息相關的變異,會跟隨著課題不同而不同。
而這五個大步驟之中,第一步的時間改進則依賴于實驗設計的改良以及試劑的研發。而后續步驟則需要依賴于算法的改進和計算設備的提升,在本文中將會介紹這些分析的常用軟件和算法,并指出這些軟件均可以通過對GPU和并行算法的優化進行提速。比如Carpi等人利用合理實驗設計和GPU,使得惡性瘧原蟲在計算成本減少4.6倍的同時整體分析速度提升27倍。
二
測序數據預處理
2.1
利用GPU獲得堿基序列
將測序過程中產生的電信號轉化為堿基的步驟被稱為 Basecalling。以 ONT測序為例,如圖 4所示,當核酸分子通過芯片上納米孔時,生成不斷變化的電信號,測序儀記錄下電信號,并轉化為堿基。
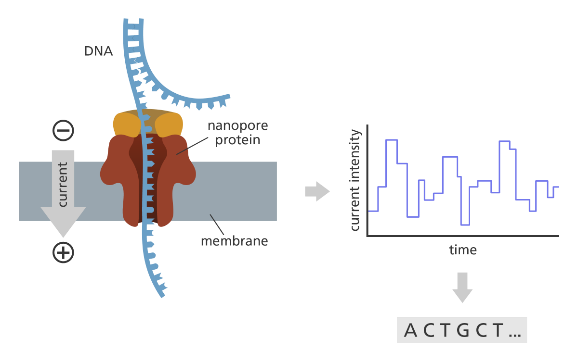
圖 4 ONT Basecalling 過程示意圖
圖片來源于What is Oxford Nanopore Technology (ONT)sequencing?,https://www.yourgenome.org/facts/what-is-oxford-nanopore-technology-ont-sequencing/
由于堿基通過納米孔的速度并不勻速,電流信號與預測得到的堿基單調但非對齊,為了解決這個問題,ONT判斷堿基的序列則是通過語音識別中常用的 CTC 作為損失函數來實現。ONT開源Basecalling 軟件Dorado 的r10.4.1系列模型中,模型分為編碼器和解碼器兩個部分,解碼器的模型如圖 3所示,其接續線性條件隨機場(CRF);解碼器則是用Viterbi算法進行的CRF解碼。Dorado對每一個測序儀器構建Fast、Hac 和 Sup 三種模型,模型依次增大且得到的堿基結果逐漸準確。r10.4.1模型中,在Fast 模式下,解碼器進入到線性CRF層的特征向量為96,狀態長度為3;Hac模式為特征向量128,狀態長度為4,而Sup模式特征向量達到了256,狀態長度為5。在 Fast模型下GPU加速效果可以達到60倍以上。

圖 5 Dorado r10.4.1 Fast模式的模型示意圖
2.2
DeepConsensus深度學習修正測序錯誤
由于PacBio會有完全隨機的錯誤發生(主要為短片段的插入與缺失),PacBio使用同一個零模波導孔內SMRT 圈多次測序得到多條Subreads序列的方法去除測序錯誤,矯正得到一個低錯誤率高質量值的序列,平均SMRT 圈的測序圈數決定了測序的時間。PacBio Revio測序儀使用CCS 和DeepConsensus 模型來矯正測序錯誤獲得高質量的序列。CCS基于隱馬爾可夫模型,其GPU版本可加速至12倍。DeepConsensus是一個編碼器-only的Transformer模型,與使用CCS的結果相比,DeepConsensus能夠得到更準確的多聚核苷酸位點。
DeepConsensus將測序得到的Reads切割,每份 100bp,每一份向量包含了序列、鏈方向、脈沖寬度、脈沖間隔以及信噪比等信息。通過一個6個自注意力層的編碼器后,使用以Softmax作為激活函數的前饋神經網絡解碼得到真實的序列。由于GPU在神經網絡訓練和推理上的優勢,相比于CPU版本的DeepConsensus,GPU加速效果能夠達到 3.3倍以上。
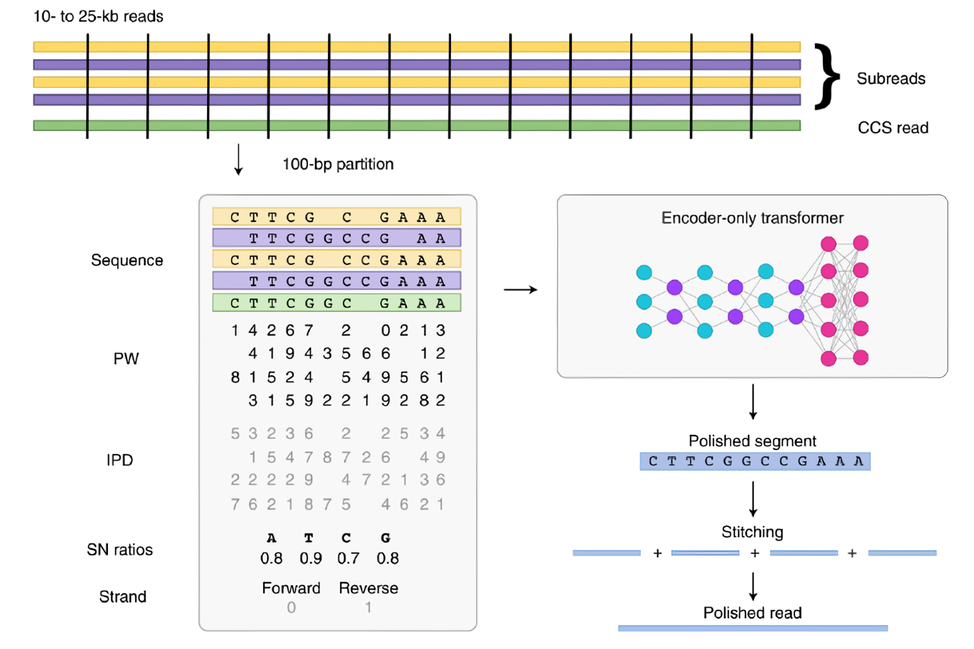
圖 6 DeepConsensus 流程示意圖
圖片來源于Baid et al. DeepConsensus improves the accuracy of sequences with a gap-aware sequence transformer. DOI: 10.1038/s41587-022-01435-7
三
測序序列比對
兩條序列比對問題是序列比對的基礎,以圖7所示,序列1 GACTTTCC 與 序列2 TAGACTACC 進行比對得到比對表。兩條DNA序列的比對通常有四種狀態:插入、缺失、匹配和錯配,而匹配和錯配往往會在比對問題中合并為同一個狀態。

圖 7 seq1與seq2的序列比對示意圖
Smith-Waterman算法和 Needleman-Wunsch算法是序列比對中常用的算法,其中Smith-Waterman算法用于局部比對而Needleman-Wunsch算法應用于全局序列比對。而實際案例中,由于參考基因組的序列長,比如人的參考序列總長度在3Gb,一次測序深度10x測序所得Reads數目在2百萬左右,直接使用局部比對的Smith-Waterman算法在時間成本上是不可接受的,于是有兩種加速方案:第一種為各測序所得序列間并行運算,另一種則是對現有算法進行改進,Minimap2等基Needleman-Wunsch的比對軟件便應運而生。在本節中首先介紹Smith-Waterman與Needleman-Wunsch的動態規劃算法與其GPU加速情況,然后介紹Minimap2特殊的GPU加速。
3.1
Smith-Waterman與Needleman-Wunsch 算法與GPU加速
兩個算法擁有相同的步驟:首先初始化得分矩陣,根據相似性矩陣和罰分矩陣填滿表格,后根據分數回溯得到比對結果。如圖 8所示,Gotoh提出的比對方式具體如下:
01
確定罰分矩陣,罰分矩陣定義了堿基匹配的獎勵分s、開啟間隔區域(插入或者缺失)的懲罰分q和持續間隔e的懲罰分。這一步往往根據測序的特性進行確定。
02
根據罰分矩陣來初始化打分矩陣,通常會將分數存儲在三個打分矩陣 H、E和F中,分別代表著匹配分數、插入分數和缺失分數。Smith-Waterman算法中,打分矩陣H、E 和F的首列與首行初始化為0;而Needleman-Wunsch算法中,打分矩陣F首列初始化公式為F0,j=F0,j-1-e,打分矩陣E首行初始化公式為Ei,0=Ei-1,0-e,打分矩陣H的首列和首行初始化為Hi,j=max{Ei,j,Fi,j}
03
計算打分矩陣H算法中的每一個位置的分數Hi,j 。Smith-Waterman 算法中Hi,j=max{Hi-1,j-1+s,Ei,j,Fi,j,0} ,而Needleman-Wunsch算法中,Hi,j=max?{Hi-1,j-1+s,Ei,j,Fi,j},其中 Ei,j=max?{Hi-1,j-q,Ei-1,j}-e,Fi,j=max?{Hi,j-1-q,Fi,j-1}-e。
當打分矩陣H、E和F全部填寫完成后,需要回溯來獲取最優的比對序列。Smith-Waterman算法從打分矩陣中的最大值開始回溯直到Hi,j=0為止;而Needleman-Wunsch算法從打分矩陣H右下角開始回溯,直到左上角為止。回溯路徑即為比對路徑。此時對于長度M和長度N的序列對而言,時間復雜度為O(MN)。
Myers提出了Bit-Vector加速該算法,打分矩陣Hi,j中的依賴于Hi-1,j-1、Hi-1,j和Hi,j-1,如果以列向量的角度看的話,如圖 9所示,那么Hj僅依賴于Hj-1。該算法將計算打分矩陣Hi,j變為計算行差值Δhi,j與列差值Δvi,j。將時間復雜度降為O(N)。
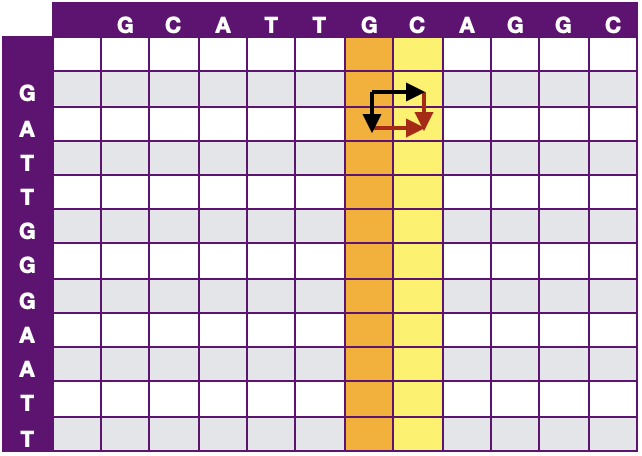
圖 9 Bit-Vector 加速示意圖
Siriwardena等人提出了另一個加速方案,如圖 10所示,次對角線上的各個元素沒有相互依賴,故拓展到次對角線的Block沒有依賴,因此通過各個Block之間的并行計算進行GPU加速。該方案運行時間與比對序列長度成非線性正相關,在如人染色體的數量級上能夠達到40倍的加速效果。
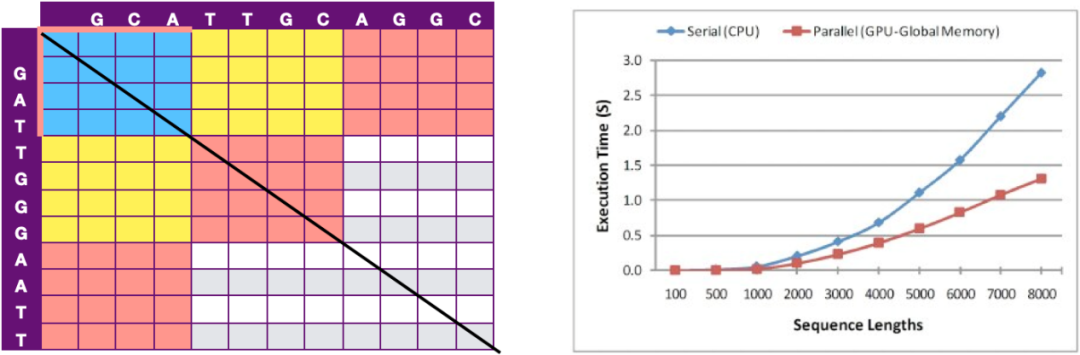
圖 10 次對角線分塊加速示意圖
與CPU 和 GPU執行時間比較圖
除了上述提到的方案以外,使用動態規劃算法的時候需要頻繁用到max、min等計算,GPU同時采用特殊的硬件指令來加速動態規劃算法,預計可以加速7倍。
3.2
比對軟件的新星Minimap2的流程與GPU加速
為了解決Smith-Waterman局部比對在大規模數據中耗時長的問題,Li開發了Minimap2 軟件極大減少了比對的時間。Minimap2 是在比對領域較新且廣泛使用的軟件,相比于其它軟件如BWA-mem、STAR等有特定使用場景的限制,Minimap2的使用范圍比較廣泛,其適用于二代的DNA短讀長測序數據以及三代DNA長讀長測序數據,也可以被應用RNA-seq這種有大間隔的數據。如圖11所示,Minimap2將測序得到的 Reads與參考基因組的比對的過程主要分為 Seeding、Chaining 和 Alignment 三個步驟。
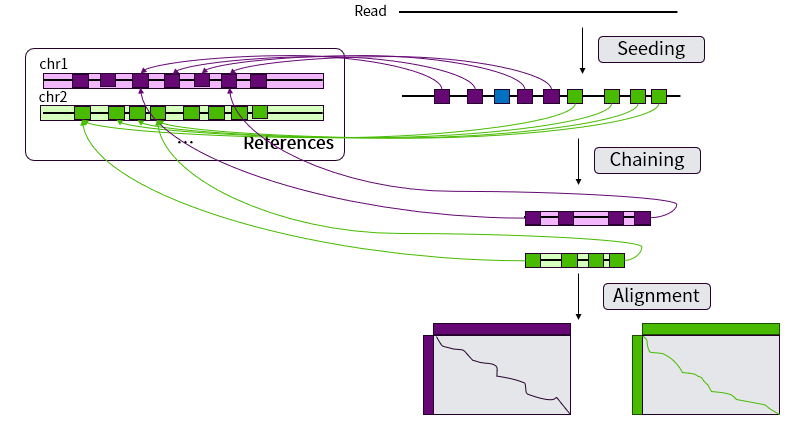
圖 11 Minimap2 流程示意圖
01
Seeding這一步快速定位參考基因組與Read之間完全匹配,無插入與缺失的短序列,得到若干有匹配位置信息的錨點(Anchors)。錨點使用A(x,y,w)表示,意味著參考基因組的坐標[x-w+1, x]與Read的坐標[y-w+1, y]完全匹配,每一條Read會得到若干個錨點。
02
Chaining這一步則是將Seeding 這步得到的錨點連起來,確定這條Read在參考基因組上的真實位置。在 Minimap2中,Chaining是用一個一維的動態規劃算法計算f 解決的,將所有錨點根據參考基因組的位置坐標x進行排序,第 i個anchor的分數f(i)可以由公式 f(i)=max?{max?{f(j)+α(j,i)-β(j,i),wi}(i>j≥1)}, 計算得到,其中 α(j,i)=min?{min??{yi-yj,xi-xj},wi} 作為獎勵分。為兩個錨點之間相同的長度;β(j,i)=γc ((yi-yj )-(xi-xj )),作為兩個錨點之間發生插入或者缺失間隔的罰分,γc 是一個與間隔長度相關的函數,可以調整 γc 以適應不同測序類型。Minimap2也會設置一個最長間隔G,當max?{yi-yj,xi-xj}>G時,罰分β(j,i)=∞。若有N個錨點,那么直接計算最大f時間復雜度為O(N2),基于大部分f(i) 的最大值在i附近,故只會計算h次,于是Minimap2將時間控制在O(hN)內。對于i每次得到f后,會回溯到 j 直到f(j)=wj ,此時將回溯過程中的錨點記為“已用”,得到一條鏈,每一個鏈根據匹配長度和間隔長度計算比對分數S。當我們將所有的錨點標記為“已用”后,可以得到所有鏈和其分數S,分數最高的鏈便是得到的最優鏈。
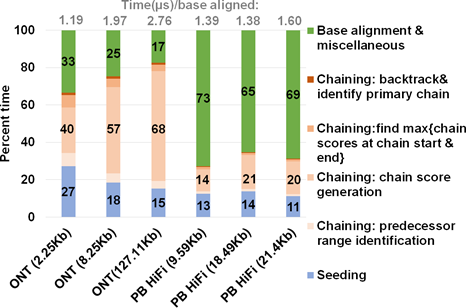
圖 12 Minimap2不同步驟所花時間比例圖。圖片來源于Sadasivan et al. Accelerating Minimap2 for Accurate Long Read Alignment on GPUs. DOI: 10.26502/jbb.2642-
03
Alignment 這一步,則是將測序得到的 Read 與 Chaining 這一步得到的鏈序列進行 base to base 的全局比對。這一步則可以用上一小節所提到的Needleman-Wunsch算法完成。
Minimap2的耗時時間如圖 12所示,Chaining和 Alignment 是耗時最長的時間。因此提升 Chaining 和 Alignment步驟的速度是提升 Miniamp2 運行速度的關鍵。
Sadasivan 等人提出了Minimap2 Chaining步驟的并行方案。如圖 13所示,他們首先將 Minimap2 的Chaining步驟的逆向搜索變成順向搜索,并且對于每一個i錨點并行計算其順向的 >i 錨點是否為最大分數。GPU通過這種方式并行即可以將Minimap2提速3.8倍左右。
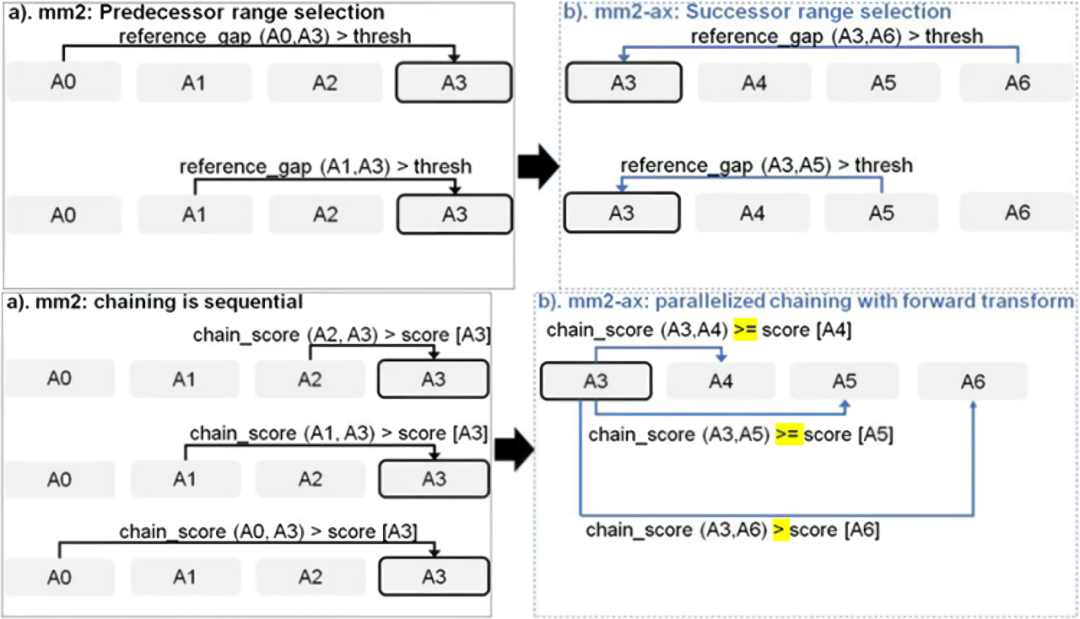
圖 13 mm2-ax 加速 Minimap2 Chaining 的方案
圖片來源于Sadasivan et al. Accelerating Minimap2 for Accurate Long Read Alignment on GPUs. DOI: 10.26502/jbb.2642-91280067
四
變異檢測
4.1
SNV與InDel的檢測
在生物學上,通常需要關注的是測序得到的變異。單堿基變異(SNV)是常見的一種變異方式,SNV 是單堿基的變異,堿基的突變有時候會改變氨基酸,或者使得轉錄提前終止。SNV的檢測主要分為兩個步驟:1. 確定并過濾變異位置; 2. 確定基因型。在本節中將介紹兩種軟件,一種是以 HaplotypeCaller和MuTect2為代表的基于傳統概率學模型建模的軟件,另一種則是以DeepVariant為代表的利用深度學習進行檢測的軟件。
4.1.1
概率模型 HaplotypeCaller / MuTect2檢測流程
HaplotypeCaller 與 MuTect2 是 GATK 中重要的 SNV 與 InDel Calling 的軟件。兩款軟件在一定程度上擁有相同的原理,在概率模型上的區別,導致HaplotypeCaller 主要用于性細胞變異的檢測,而MuTect2 更適用于體細胞變異的檢測。如圖 14兩款軟件都在確定變異區間后,利用局部組裝得到的單倍型并將測序得到的Reads用Smith-Waterman算法比對到單倍型中來減少測序、比對等錯誤信息,通過 PairHMM 來得到每一個單倍型的似然值,然后根據各自的概率模型獲得最終的基因型。
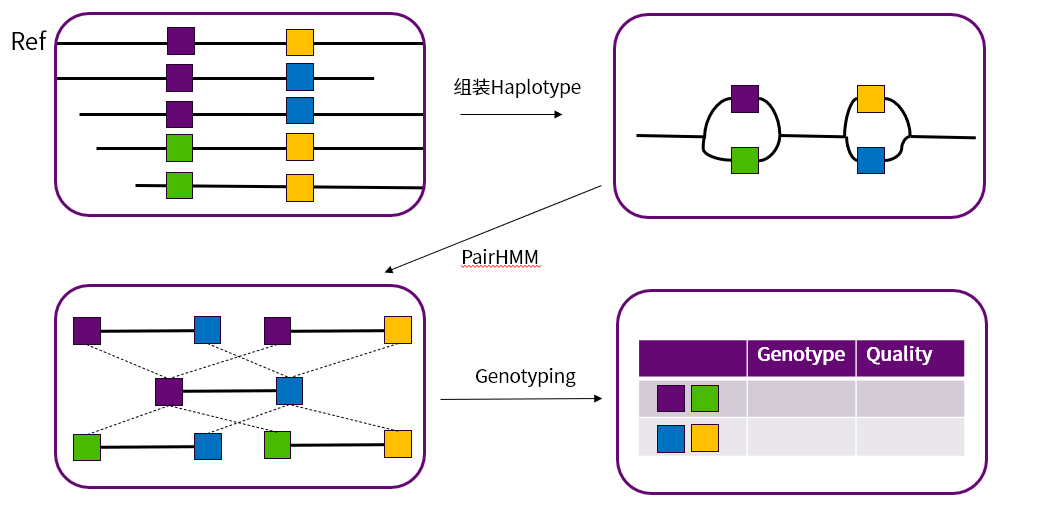
圖 14 HaplotypeCaller與MuTect2 的通用流程示意圖
每一個單倍型的似然值時HaplotypeCaller和MuTect2的關鍵步驟,即求單倍型的集合H對于所有測序得到的Reads的集合R的比對結果集合A的概率,即 P(R│H)=∑AP(R,A|H),那么只需要求解每一個比對的概率即可。其中我們已經看到了重新比對中Smith-Waterman的GPU加速方案,HaplotypeCaller中的PairHMM這一步亦可以用GPU并行加速。
PairHMM 的過程與前述的比對算法相似,定義了三個狀態: 匹配M, 插入I和缺失D。如圖15所示,三個狀態會發生轉移, M→I或者 M→D,即從匹配狀態轉化為插入狀態或者缺失狀態概率為δ、 I→M 從插入狀態結束間隔變回匹配狀態概率為ε、D→M從缺失狀態結束間隔變回匹配狀態概率為ε,也會發生持續 M→M 即延續匹配狀態概率為1-2δ、I→I 延續插入狀態概率為1-ε和D→D 延續缺失狀態,概率為1-ε。基于生物的序列特征,往往1-ε<δ?1-2δ,即維持間隔的概率小于發生間隔的概率,遠小于匹配的概率。
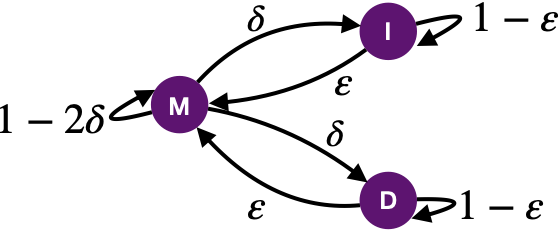
圖 15 PairHMM 狀態轉化示意圖
計算時定義了三個狀態分別定義了一個矩陣 M、I和D。Mi,j表示測序序列i與參考基因組j位置匹配的概率,同理 Ii,j和Di,j分別代表著發生插入的概率和缺失的概率。矩陣M可以通過公式 Mi,j=S(Mi-1,j-1 (1-2δ)+Ii-1,j ε+Di,j-1 ε) 計算而得,其中S與堿基的質量值錯配概率等有關;矩陣I可以通過公式Ii,j=Mi-1,j δ+Ii-1,j (1-ε) 得到;矩陣D可以通過公式Di,j=Mi,j-1 δ+Di,j-1 (1-ε) 得到;當計算完矩陣M、I后,將M與I的最后一列相加即可得到似然值。
PairHMM是檢測流程中最密集的部分,因此提升 PairHMM的速度就是提升 HaplotypeCaller/MuTect2 的關鍵,可以發現PairHMM的分數矩陣計算過程與Smith-Waterman的打分矩陣過程相似,于是Li 等人參考Smith-Waterman的GPU加速方案提出的基于 GPU的 PairHMM 比CPU版本加速 783 倍。
4.1.2
DeepVariant深度學習檢測SNV
HaplotypeCaller/Mutect2 傳統的概率模型算法存在兩個問題:
1
由于不同的測序技術或者物種不同,HaplotypeCaller所需要的往往先驗參數不同。比如PacBio測序中,插入發生的概率大于缺失發生的概率,遠大于傳統的 Illumina 二代的概率,于是 HaplotypeCaller 需要根據測序技術的更迭不斷嘗試獲得最優的先驗參數。
2
傳統概率模型的建模中假設了SNV在基因組發生的概率相同。而由于自然選擇的存在,SNV的發生概率在基因組中并非均勻。在生物中,往往位于內含子區域的變異發生概率會大于外顯子區域概率,而管家基因內變異發生概率會遠小于其它基因變異發生概率。
為了解決這個問題,DeepVariant與 HaplotypeCaller 不同,利用深度學習的方法代替傳統的概率模型。如圖 16所示,在確定變異的位置后,DeepVariant將覆蓋該位置的 Reads 提取出來,組成一個 PileUp 的圖像,圖像包含了該位點上下游100bp的序列、質量值、Reads 鏈、比對質量、變異支持及覆蓋情況等信息,這些信息將起到與 HaplotypeCaller 組裝單倍型的相似作用。

圖 16 DeepVariant MakeExample 所以生成的典型圖片,分別為純合SNV變異、雜合SNV變異和純合未變異。圖片來源于Looking Through DeepVariant’s Eyes. https://google.github.io/deepvariant/posts/2020-02-20-looking-through-deepvariants-eyes/
得到圖片后,DeepVariant 使用 CNN 將 PileUp 得到的圖片推理直接獲得該位點基因型的似然值,從而判斷是否發生變異以及變異的基因型。
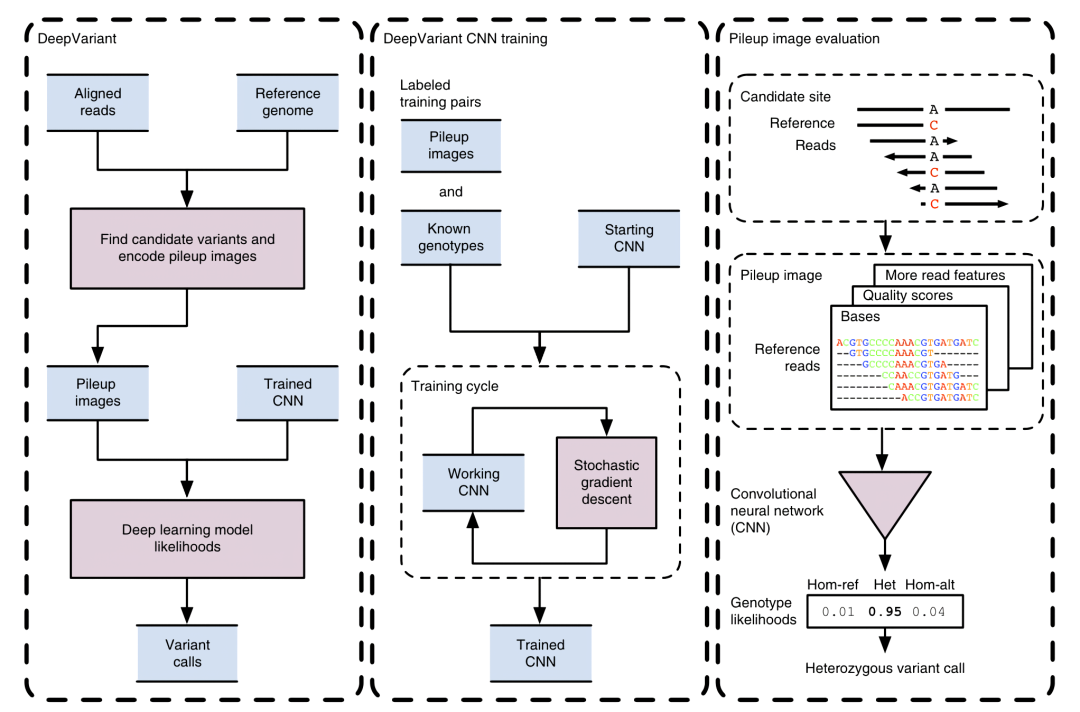
圖 17 DeepVariant流程示意圖
圖片來源于Poplin et al. A universal SNP and small-indel variant caller using deep neural networks. DOI: 10.1038/nbt.4235
DeepVariant 在測序技術和物種都具有泛化性,用戶可以通過兩個個體的測序數據構建純合和雜合的“銀標準”對DeepVariant的模型進行微調,使DeepVariant適合不同的測序技術(如 Illumina、PacBio HiFi、 ONT R10.4.1等等)。另一方面,DeepVariant 也通過對輸入或者輸出改造,已經衍生出應用于其他場景的模型,比如適用于家系的 DeepTrio,或者利用腫瘤樣本-正常細胞樣本對檢測體細胞變異的 VarNet。如圖 18所示,DeepVariant 在 GPU 的幫助下可以顯著減少 Variants Calling 這個步驟。
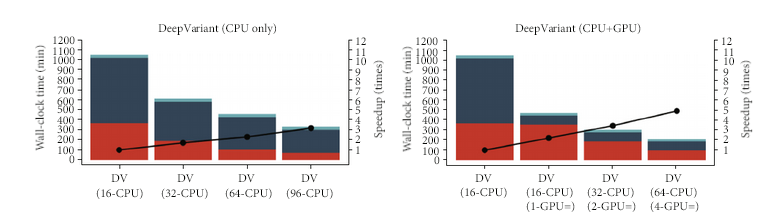
圖 18 DeepVariant不同硬件不同環節時間統計圖。
圖片來源于 Huang et al. DeepVariant-on-Spark: Small-Scale Genome Analysis Using a Cloud-Based Computing Framework. DOI: 10.1155/2020/7231205
4.2
結構變異檢測
結構變異通常包括了插入、刪除、串聯重復、染色體倒位、染色體易位、拷貝數變異和復雜的復合結構變異等變異。結構變異涉及到>50bp的序列,與SNV或小片段的InDel相比,一旦變異發生通常更不可逆。結構變異的檢測有著重要的意義:
1
由于不可逆的特性,結構變異往往是人種區別、物種分化的關鍵。
2
稀有的結構變異往往與疾病或者癌癥發生關聯。
3
在腫瘤細胞中變異會出現大量的高密度的異常的結構變異,甚至會出現染色體碎裂的現象。
結構變異的檢測的方法如圖 19所示,包括了基于雙端測序方向的Read Pair、基于測序深度變化的 Read Depth、基于比對時異常比對的Split Reads 和將測序得到的 Reads組裝與參考基因組比對的 Assembly。目前短序列有Manta、Delly,而長序列有 Sniffles2、CuteSV等軟件,基于比對時異常比對的Split Reads是主流的方法。
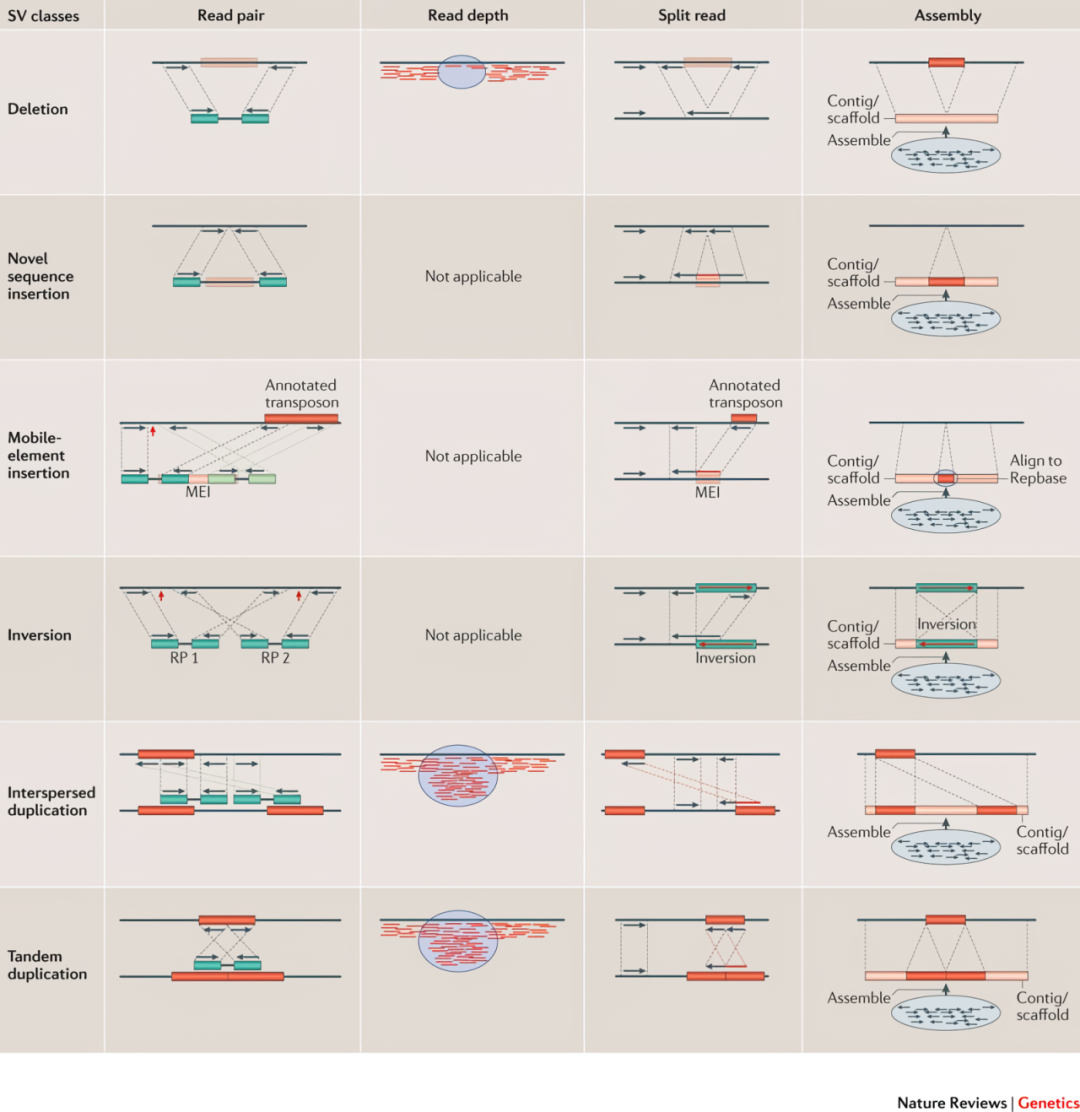
圖 19 結構變異檢測的常用方法
圖片來源于Alkan et al. Genome structural variation discovery and genotyping. DOI: 10.1038/nrg2958
4.2.1
SVision深度學習模型
如 Sniffles2、Manta等結構變異軟件,過濾得到比對異常的Reads,將比對異常點根據其在參考基因組的位置、異常的長度等方式進行聚類,每一個類即獲得一個結構變異位點。但是這缺少了發現復合結構變異的能力。通常復合結構變異檢測都依賴于現有的模式,如易位后缺失等,缺乏一個統一的結構。
SVision 便是為了解決這個問題而出現的模型31。如圖 20所示,SVision包含了編碼器、tMOR和解釋器。編碼器將測序的序列轉化為圖片,其識別支持變異序列和參考基因組序列的堿基通過VAR-REF和REF-REF圖像,從VAR-REF中去除REF-REF中已有的重復序列,以提高后續分析的準確度。在tMOR步驟中,SVison將包含多個復合結構變異斷點的圖像通過5層卷積層、1層展平層和2層全連接層的卷積神經網絡進行預測發生變異的概率。最后,解釋器描述了不同的復合結構變異。由于這是一個卷積神經網絡的模型,GPU的矩陣乘法上的優勢能夠比CPU版本的SVision快1.8倍。
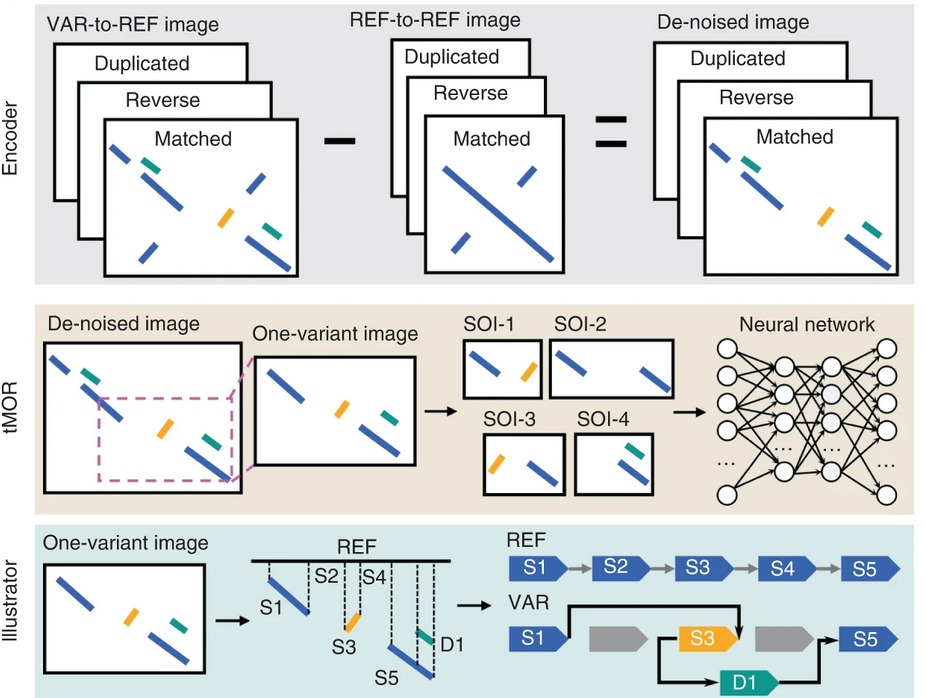
圖 20 SVision 流程示意圖
圖片來源于 Lin et al. SVision: a deep learning approach to resolve complex structural variants. DOI: 10.1038/s41592-022-01609-w
四
總結與展望
變異檢測是定位功能基因或疾病基因的基礎。如表格 1所示,從測序數據的產生、預處理、基因組的比對、變異檢測都可以使用GPU加速來提升檢測的速度。高速分析能夠節省測序的時間成本,從而降低測序價格和分析成本。這可以讓研究人員更愿意進行測序來建立與完善疾病腫瘤相關的數據庫,為將來攻克疾病與腫瘤打下基礎。
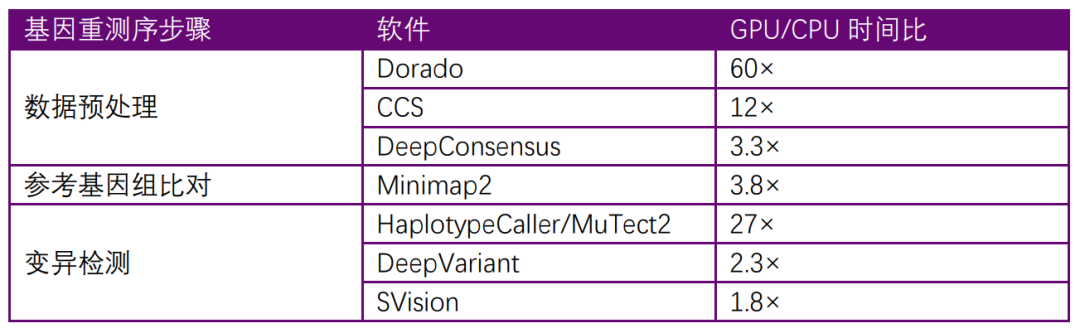
表格 1 文中所提的軟件GPU與CPU運行時間比
可以看到,在目前基因測序領域,傳統軟件算法因為設備局限性,在面對復雜的生物系統時,選擇使用簡化的概率模型建模來犧牲準確度而獲得更快的速度。而隨著GPU算力的進步,越來越多研究人員選擇深度學習或者大模型來得到更精確的結果。除了文中所提到的數據預處理、基因組比對和變異檢測步驟以外,在后續分析中,機器學習也起到了更重要的作用,
比如用線性模型建模關聯樣本表型與基因型的GWAS可以使用 DeepNull 得到更好的關聯結果。
我們預計,隨著機器學習算法和GPU設備的發展,快速而準確的基因測序可以幫助研發人員設計靶向藥物,幫助普通民眾更早發現潛在風險,幫助醫生更準確地對病情進行診斷治療,精準醫療未來可期。
名詞列表
責任編輯:彭菁
-
傳感器
+關注
關注
2548文章
50740瀏覽量
752140 -
gpu
+關注
關注
28文章
4703瀏覽量
128725 -
基因
+關注
關注
0文章
95瀏覽量
17195
發布評論請先 登錄
相關推薦
Xilinx FPGA在基因組測序中的優勢
全基因組數據CNV分析簡介 精選資料分享
全基因組測序的優勢 精選資料分享
什么是基因組序列數據庫
全國首個“城區全民基因組測序項目”在深圳啟動,基因科技進入臨床應用
FPGA能在實時基因組測序計算中大顯身手,大大縮短時間
Clay Breshears博士討論基因組測序和生物信息學
國產芯片助力全球首次實現手機個人全基因組測序分析
國產芯片助力全球首次實現手機個人全基因組測序分析
AI加速推動醫療個體化轉型 基因組學將有望成為未來發展主流
GPU助力基因組重測序分析





 工商網監
工商網監
評論