") 字節(jié)跳動(dòng)李航:AI for Science的一些探索和進(jìn)展
字節(jié)跳動(dòng)李航:AI for Science的一些探索和進(jìn)展
近年,人工智能的各個(gè)領(lǐng)域,包括自然語言處理、計(jì)算機(jī)視覺、語音處理,借助深度學(xué)習(xí)的強(qiáng)大威力,都取得了令人嘆為觀止的巨大進(jìn)步。將深度學(xué)習(xí)技術(shù)應(yīng)用于傳統(tǒng)的科學(xué)領(lǐng)域,如物理、化學(xué)、生物、醫(yī)學(xué),即所謂的 AI for Science(科學(xué)智能),作為一個(gè)新的交叉學(xué)科,也逐漸興起,孕育著巨大的潛力,受到廣泛的關(guān)注。
ByteDance Research 也在進(jìn)行 AI for Science 的研究,包括機(jī)器學(xué)習(xí)與量子化學(xué)、大規(guī)模量子化學(xué)計(jì)算、AI 制藥等領(lǐng)域一些問題的研究,希望跟業(yè)界一起推動(dòng)領(lǐng)域的發(fā)展。本文簡要介紹我們這兩年來取得的一些進(jìn)展。也拋磚引玉,希望與業(yè)界進(jìn)行更多的交流和合作。
在機(jī)器學(xué)習(xí)和量子化學(xué)方向,我們提出的 LapNet 算法,比有代表性的 FermiNet 模型訓(xùn)練速度提高了 10 倍,能計(jì)算的化學(xué)體系的規(guī)模和精度目前是領(lǐng)域最大的。
在大規(guī)模量子化學(xué)計(jì)算方向,我們開發(fā)了 Periodic DMET 算法,使用經(jīng)典和量子混合計(jì)算機(jī)(實(shí)際是在經(jīng)典計(jì)算機(jī)上的模擬),用于周期性體系的計(jì)算,只用 20 個(gè)量子比特就達(dá)到了之前方法用近萬個(gè)量子比特才能達(dá)到的精度。
在 AI 制藥方向,我們開發(fā)的 LM-Design 模型,利用大量蛋白質(zhì)序列數(shù)據(jù),以及一定數(shù)量的蛋白質(zhì)結(jié)構(gòu)和序列對(duì)應(yīng)數(shù)據(jù),學(xué)習(xí)從蛋白質(zhì)結(jié)構(gòu)到序列轉(zhuǎn)換的模型,達(dá)到了目前蛋白質(zhì)序列設(shè)計(jì)的最高精度。
機(jī)器學(xué)習(xí)與量子化學(xué)
物理學(xué)家狄拉克曾說:對(duì)大部分物理學(xué)和整個(gè)化學(xué),進(jìn)行數(shù)學(xué)建模所需要的基本定律已完全清楚,困難只在于這些定律的應(yīng)用,得到的方程一般都太復(fù)雜而無法求解。
量子化學(xué)是根據(jù)量子力學(xué)的原理研究化學(xué)現(xiàn)象的學(xué)科。其重要的問題是用計(jì)算的方法求解分子或周期性體系(如固體)的電子薛定諤方程,從而推算出分子或周期性體系的基態(tài)能量、電極性等特性。是所謂的從頭計(jì)算(ab initio)問題。傳統(tǒng)的方法有密度泛函理論 DFT、耦合簇 CCSD 等。要么計(jì)算的精度不夠高,要么能計(jì)算的規(guī)模不夠大。
近年,用機(jī)器學(xué)習(xí)的方法解決從頭計(jì)算問題成為倍受關(guān)注的新方向。其基本想法是借助深度學(xué)習(xí)強(qiáng)大的表示和學(xué)習(xí)能力,大幅提升從頭計(jì)算的精度和規(guī)模。其中的一個(gè)路徑是 NN-VMC(Neural Network based Variational Monte Carlo) 。用神經(jīng)網(wǎng)絡(luò)近似薛定諤方程的波函數(shù),通過隨機(jī)采樣的方式獲得體系中電子在空間中的樣本,這樣可以計(jì)算基于薛定諤方程的整個(gè)體系的能量。通過最小化能量的上界,優(yōu)化神經(jīng)網(wǎng)絡(luò)的參數(shù),不斷迭代,最后得到近似最優(yōu)的神經(jīng)網(wǎng)絡(luò)(波函數(shù)),以及體系的近似基態(tài)能量(最小能量)。(注:波函數(shù)的平方是電子在空間出現(xiàn)的概率密度函數(shù),有了波函數(shù),就可以進(jìn)行電子在空間中的隨機(jī)采樣。)圖 1 顯示 NN-VMC 的基本原理。其核心問題是如何設(shè)計(jì)神經(jīng)網(wǎng)絡(luò)和學(xué)習(xí)算法。
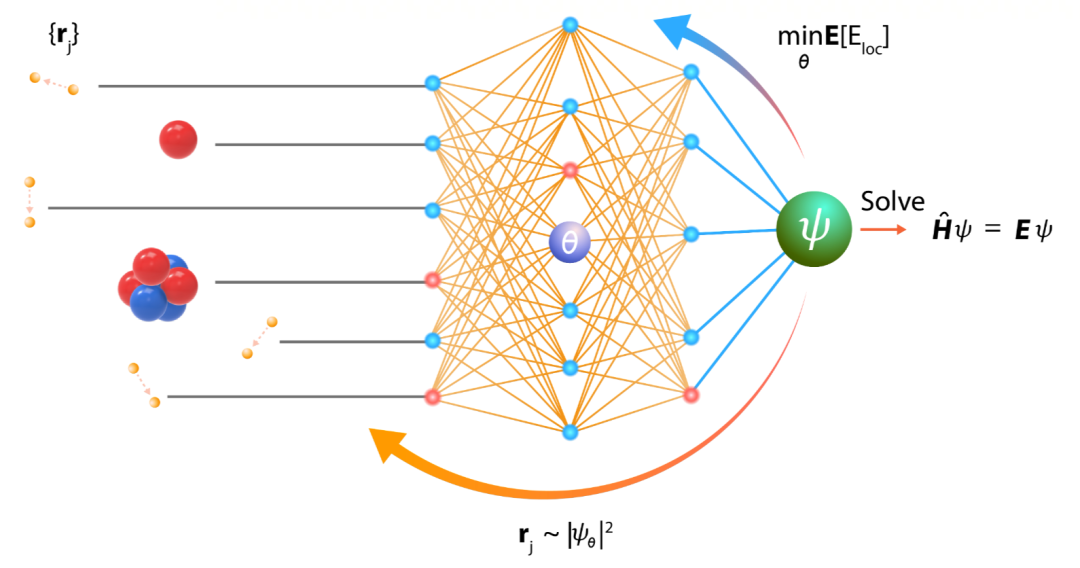
圖 1. NN-VMC 方法的基本原理
NN-VMC 中有代表性的方法是 DeepMind 和 ICL 于 2019 年提出的 FermiNet。之后一些研究機(jī)構(gòu)又提出了一些新的方法。ByteDance Research 從 2021 年起,與北京大學(xué)合作,進(jìn)行了一系列相關(guān)研究,提出了幾個(gè)新的方法。下面對(duì)這些方法做一簡單介紹。
NN-VMC+ECP,是我們開發(fā)的結(jié)合 NN-VMC 和贗勢 ECP(Effective Core Potential)的方法 [1],可以進(jìn)一步提高計(jì)算的效率和體系的規(guī)模。計(jì)算化學(xué)體系的特性時(shí),往往只需要關(guān)注原子中外側(cè)軌道的電子。將原子中內(nèi)側(cè)軌道的電子的勢能用定量表示,就可以大幅減少所需要的計(jì)算量。我們將 ECP 技巧應(yīng)用于 NN-VMC,得到了這個(gè)新方法,取得了很好的效果。
NN-DMC,是我們提出的將神經(jīng)網(wǎng)絡(luò)和擴(kuò)散蒙特卡洛法 DMC(Diffusoon Monte Carlo)結(jié)合的另一個(gè)方法 [2]。DMC 與 VMC 不同,不是計(jì)算體系基態(tài)能量的上界,而是使用虛時(shí)演化來計(jì)算體系的基態(tài)能量。這個(gè)方法,相比 FermiNet 等已有方法也能大幅提高計(jì)算的精度和規(guī)模。
最近開發(fā)的 LapNet 也是一種 NN-VMC 方法 [3],特點(diǎn)是在神經(jīng)網(wǎng)絡(luò)學(xué)習(xí)時(shí)使用前向拉普拉斯算子( Forward Laplacian)。基于薛定諤方程計(jì)算體系的能量上界的過程中,需要計(jì)算哈密頓算子,包括其中的動(dòng)能部分。之前的方法都是通過計(jì)算相關(guān)的黑塞矩陣的方式計(jì)算動(dòng)能,其算法復(fù)雜度高,成為學(xué)習(xí)的一個(gè)瓶頸。LapNet 在學(xué)習(xí)的前向傳播中,通過拉普拉斯算子的計(jì)算,直接計(jì)算動(dòng)能,以及哈密頓算子,從而省去了黑塞矩陣的計(jì)算。這樣可以大幅提高學(xué)習(xí)的計(jì)算效率。相比 FermiNet,LapNet 有平均 10 倍左右的加速。
ECP、DMC 和 Forward Laplace 屬于三種不同的技術(shù)改進(jìn)(簡化勢能計(jì)算、優(yōu)化采樣,提高計(jì)算效率),三個(gè)技術(shù)結(jié)合起來原理上可以更大程度上提高計(jì)算規(guī)模,也是我們正在嘗試的方法。另外,我們還將 NN-VMC 方法應(yīng)用于固體的薛定諤方程求解 [4],分子體系的力場 [5]、電極化計(jì)算 [6] 等問題,證明了 NN-VMC 方法的實(shí)用性。
圖 2 顯示目前 NN-VMC 方法中代表性工作的精度和規(guī)模,縱軸表示精度,圓的大小表示規(guī)模。我們提出的 LapNet 方法能夠以更高的精度計(jì)算更大的體系。最大的體系有 116 個(gè)電子。
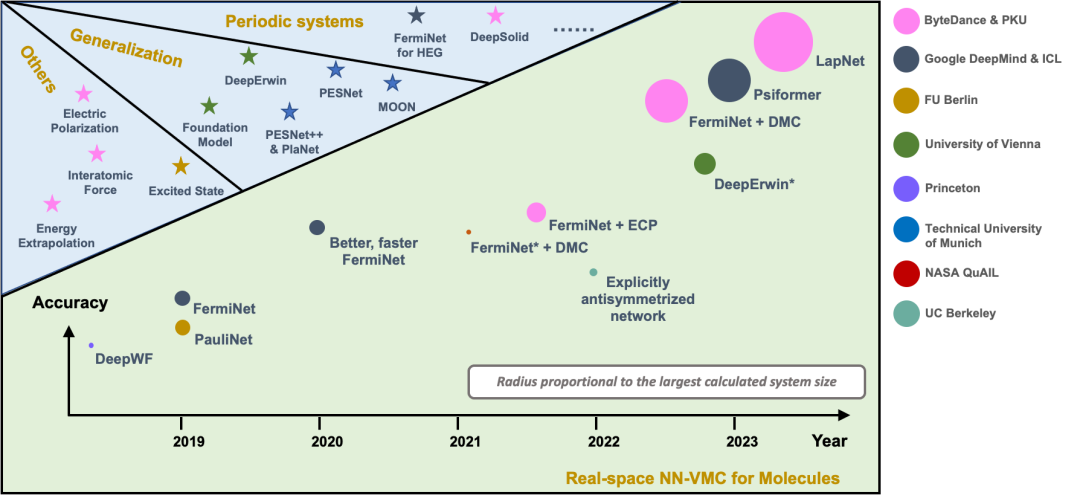
圖 2. NN-VMC 方法的規(guī)模和精度
大規(guī)模量子化學(xué)計(jì)算
通過直接求解薛定諤方程計(jì)算化學(xué)體系特性(比如基態(tài)能量)的方法能處理的規(guī)模仍然有限。量子嵌入方法(Quantum Embedding Method)被認(rèn)為是解決這個(gè)問題的一條有效路徑。基本想法是通過分而治之和多精度計(jì)算實(shí)現(xiàn)大規(guī)模化。代表性的方法有密度矩陣嵌入理論 DMET(Density Matrix Embedding Theory)。將體系劃分為若干部分(Fragment),對(duì)其中的每個(gè) Fragment 及其對(duì)應(yīng)的環(huán)境(Bath)進(jìn)行高精度計(jì)算,對(duì)其他部分進(jìn)行低精度計(jì)算。而且根據(jù)需要對(duì)每個(gè) Fragment 進(jìn)行并行處理。最后再把高精度計(jì)算的結(jié)果合并起來,不斷迭代逼近原始體系。這樣可以大幅提高可計(jì)算的體系的規(guī)模。
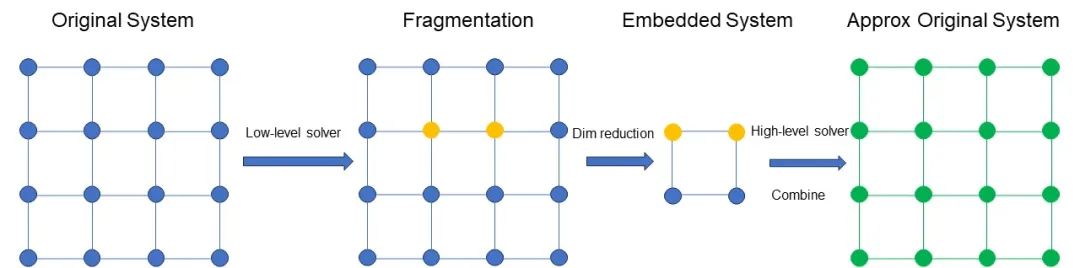
圖 3 DMET 方法的直觀解釋
圖 3 示意 DMET 方法的過程。首先對(duì)原始體系進(jìn)行劃分,得到一組 Fragment。假設(shè)圖中黃色部分是我們關(guān)注的 Fragment,例如是兩個(gè)原子。圖中藍(lán)色的部分包含環(huán)境和其他部分。對(duì)關(guān)注的 Fragment 及其環(huán)境用高精度的方法計(jì)算,例如 CCSD,對(duì)其他部分用低精度的方法計(jì)算,例如,Hartree–Fock 法。對(duì)所有的 Fragment 做同樣的并行處理。
具體算法如下。首先,通過低精度求解,得到整體(關(guān)注的 Fragment、環(huán)境和其他部分)的約化密度矩陣,這個(gè)低精度解包含參數(shù)。其次,對(duì)這個(gè)矩陣的 Fragment 及其環(huán)境進(jìn)行奇異值分解,構(gòu)建投影算符 P(該投影算符僅關(guān)注 Fragment 及其環(huán)境),利用投影算符構(gòu)建低維度的體系(圖片),并對(duì)其進(jìn)行高精度求解。之后,將所有 Fragment 的計(jì)算結(jié)果合并,作為體系整體的近似。最后,通過迭代,調(diào)節(jié)參數(shù),使得原始的低精度解逐漸逼近合并的高精度解(在 L2 norm 的意義下),直到得到最終結(jié)果。
我們基于兩種完全不同的計(jì)算范式,實(shí)現(xiàn) DMET 及其變種的 SIE,進(jìn)行大規(guī)模量子化學(xué)體系的計(jì)算。一是使用經(jīng)典計(jì)算機(jī),另一個(gè)是使用量子計(jì)算機(jī)。本文主要介紹后者的相關(guān)工作(前者的工作計(jì)劃今后有更大進(jìn)展時(shí)介紹),也稱為量子計(jì)算化學(xué)。我們考慮在量子計(jì)算機(jī)上的實(shí)現(xiàn),但只在經(jīng)典計(jì)算機(jī)上進(jìn)行模擬,希望為量子化學(xué)的發(fā)展做出貢獻(xiàn)。無論是哪種計(jì)算范式,DMET 方法使得大規(guī)模計(jì)算成為可能,我們正在嘗試努力實(shí)現(xiàn) DMET,將可計(jì)算的體系提升幾個(gè)數(shù)量級(jí)。
物理學(xué)家費(fèi)曼曾說:自然不是古典力學(xué),如果你想模擬自然,你最好用量子力學(xué)。開發(fā)量子計(jì)算技術(shù)的驅(qū)動(dòng)力就是用量子層面的計(jì)算設(shè)備模擬量子現(xiàn)象。換言之,量子化學(xué)是量子計(jì)算最合適的應(yīng)用領(lǐng)域之一。
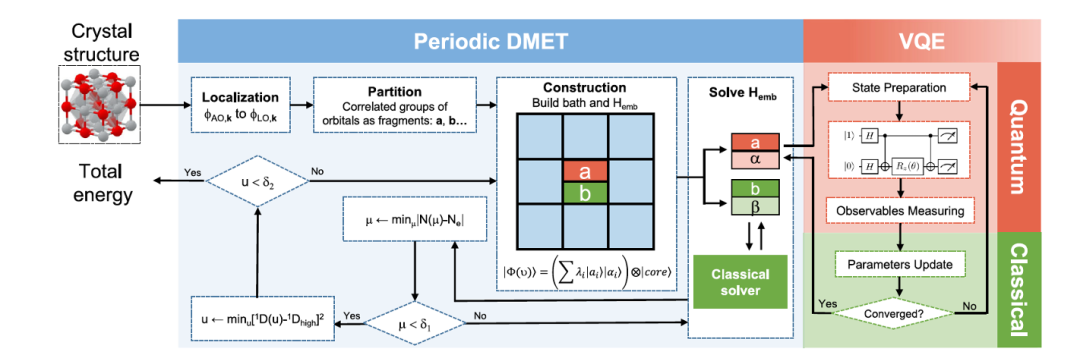
圖 4. Periodic DMET 方法的示意
我們先后開發(fā)了兩個(gè)量子計(jì)算化學(xué)方法,結(jié)合量子和經(jīng)典混合計(jì)算機(jī)和 DMET 的特點(diǎn),大幅提高了計(jì)算體系的精度和規(guī)模。基本想法是用量子計(jì)算機(jī)實(shí)現(xiàn) DMET 高精度計(jì)算的部分,用經(jīng)典計(jì)算機(jī)實(shí)現(xiàn) DMET 低精度計(jì)算的部分。DMET-ESVQE 計(jì)算分子體系 [7], Periodic DMET 計(jì)算周期性體系 [8]。前者只用 16 個(gè)量子比特,就能實(shí)現(xiàn)之前的方法用 144 個(gè)量子比特的計(jì)算。后者只用 20 個(gè)量子比特就能實(shí)現(xiàn)之前的方法用將近 1 萬個(gè)量子比特的計(jì)算。
圖 4 顯示在混合計(jì)算機(jī)上實(shí)現(xiàn)的 DMET Periodic 方法。輸入是晶體,輸出是體系的能量。首先對(duì)體系進(jìn)行劃分,然后并行地計(jì)算每一個(gè) Fragment。對(duì)關(guān)注 Fragment 及其環(huán)境在量子計(jì)算機(jī)上用 U-CCSD 求解。對(duì)其他部分在經(jīng)典計(jì)算機(jī)上用 Hartree–Fock 法求解。
量子蒙特卡羅法,包括 VMC、DMC,是量子化學(xué)的最有效的一系列算法 [9]。我們還提出了將量子計(jì)算和量子蒙特卡羅法結(jié)合的新方法。這個(gè)方法可以體現(xiàn)量子計(jì)算對(duì)量子化學(xué)的一些優(yōu)勢。具體地,量子計(jì)算可以部分解決量子蒙特卡洛法中的符號(hào)問題。
AI 制藥
使用 AI 技術(shù)輔助藥物發(fā)現(xiàn)已經(jīng)成為被業(yè)界廣為接受的新范式。近年有大量的研究,也有一些技術(shù)應(yīng)用于實(shí)際的場景。我們進(jìn)行了基于 AI 技術(shù)的藥物設(shè)計(jì)的研究和開發(fā),包括小分子藥物和大分子藥物(抗體藥物)。
小分子藥物設(shè)計(jì)過程包括蛋白質(zhì)靶點(diǎn)的發(fā)現(xiàn),小分子藥物候選的生成,候選與靶點(diǎn)(target)的親和性、候選的成藥性、無毒性等的判斷。目前有 AI 技術(shù)可以實(shí)現(xiàn)這些藥物的開發(fā)步驟。我們開發(fā)了基于機(jī)器學(xué)習(xí)的小分子藥物候選生成的方法,MARS 是利用打分函數(shù)自動(dòng)生成候選的方法 [10],DESERT 是根據(jù)靶點(diǎn)的形狀自動(dòng)生成候選的方法 [11]。
MARS 從種子分子開始,不斷編輯分子,直到最后得到最優(yōu)的小分子藥物候選。生成的過程中使用馬爾可夫鏈蒙特卡洛法(MCMC),其平穩(wěn)分布是由多個(gè)打分函數(shù)構(gòu)成的概率分布。打分函數(shù)表示小分子藥物候選的親和性、成藥性、無毒性等。建議分布表示基于圖神經(jīng)網(wǎng)絡(luò)(MPNN)進(jìn)行小分子藥物候選的編輯前后的條件概率分布。圖神經(jīng)網(wǎng)絡(luò)表示小分子化合物的分子式,結(jié)點(diǎn)是原子,邊是化學(xué)鍵。對(duì)小分子的編輯包括增加新的結(jié)點(diǎn)和刪除已有的結(jié)點(diǎn)。圖神經(jīng)網(wǎng)絡(luò)上可以預(yù)測對(duì)小分子的可能的編輯操作(增加或刪除),其參數(shù)通過學(xué)習(xí)得到。MARS 只需要有打分函數(shù)、小分子藥物的數(shù)據(jù)庫(分子式)就可以生成全新的和多樣的小分子藥物候選。目前 MARS 已經(jīng)被用于實(shí)際的小分子藥物設(shè)計(jì)工作中。
DESERT 通過兩個(gè)步驟進(jìn)行小分子藥物候選的生成。Sketching:采樣與蛋白質(zhì)靶點(diǎn)的口袋(pocket)形狀互補(bǔ)的藥物候選的形狀,Generating:基于藥物候選的形狀自動(dòng)生成藥物候選的分子式。圖 5 顯示這個(gè)過程。
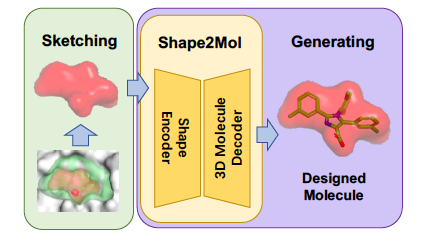
圖 5. DESERT:自動(dòng)生成小分子藥物候選
候選和靶點(diǎn)結(jié)合,其必要條件是兩者的形狀能夠進(jìn)行很好的對(duì)接。在 Sketching 階段,根據(jù)蛋白質(zhì)靶點(diǎn)的形狀,用啟發(fā)式的方法產(chǎn)生候選的形狀。在 Generating 階段,用事先學(xué)好的形狀到分子的生成模型 Shape2Mol 根據(jù)形狀自動(dòng)生成分子式。可以利用分子庫里的大量的藥物的分子式和形狀,學(xué)習(xí)這個(gè)生成模型。如圖 6 所示,Shape2Mol 中,編碼器將分子 3D 形狀進(jìn)行編碼,產(chǎn)生中間表示,解碼器根據(jù)中間表示生成分子式。3D 形狀使用體素表示,分子式使用符號(hào)序列表示。DESERT 是 2022 年小分子藥物候選生成結(jié)合性能最好的方法。
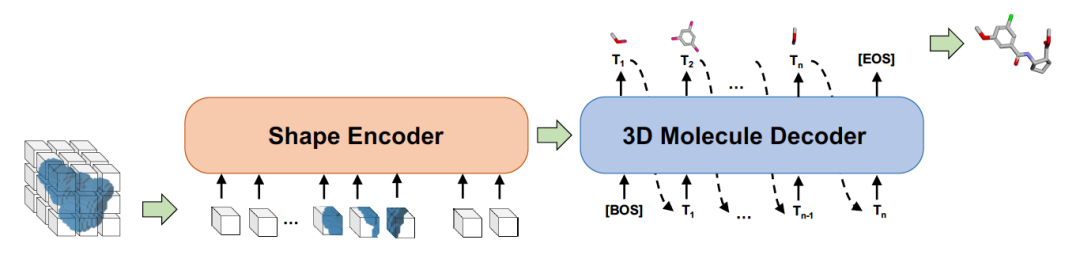
圖 6. 形狀到分子的生成模型 Shape2Mol 的示意
最近我們聚焦在大分子藥物設(shè)計(jì)上,更一般的問題是蛋白質(zhì)設(shè)計(jì)。蛋白質(zhì)設(shè)計(jì)包括抗體藥物設(shè)計(jì),多肽藥物設(shè)計(jì)等。如果知道了蛋白質(zhì)序列(氨基酸序列),就可以預(yù)測其結(jié)構(gòu),知道了蛋白質(zhì)結(jié)構(gòu)也就能預(yù)測其功能。這就是著名的 AlphaFold 做的事情。蛋白質(zhì)設(shè)計(jì)可以看作是一個(gè)反向的過程。一般從功能出發(fā)決定對(duì)應(yīng)的蛋白質(zhì)結(jié)構(gòu),再根據(jù)蛋白質(zhì)結(jié)構(gòu)決定對(duì)應(yīng)的蛋白質(zhì)序列。我們開發(fā)了從蛋白質(zhì)結(jié)構(gòu)生成蛋白質(zhì)序列的模型 LM-Design。
LM-Design 的輸入是蛋白質(zhì)結(jié)構(gòu),輸出是對(duì)應(yīng)的蛋白質(zhì)序列。LM-Design 由結(jié)構(gòu)編碼器和序列解碼器組成。其中,結(jié)構(gòu)編碼器是已訓(xùn)練好的表示蛋白質(zhì)結(jié)構(gòu)的圖神經(jīng)網(wǎng)絡(luò);序列解碼器基于已預(yù)訓(xùn)練好的大規(guī)模蛋白質(zhì)語言模型(Protein Language Model),與 BERT/Transformer Encoder 相似(使用雙向自注意力),在最后一層插入一個(gè)結(jié)構(gòu)適配器(Structural Adaptor)。結(jié)構(gòu)適配器的參數(shù)是待學(xué)習(xí)的。圖 7 給出 LM-Design 的模型架構(gòu)。
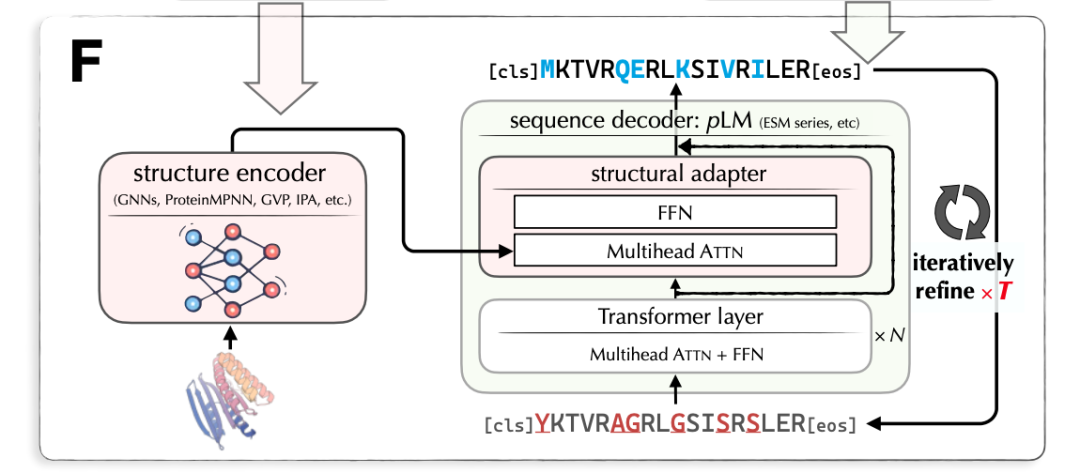
圖 7. 蛋白質(zhì)結(jié)構(gòu)到序列的生成模型 LM-Design 的架構(gòu)
LM-Design 的學(xué)習(xí)和預(yù)測是掩碼語言建模(Masked Language Modeling),與 BERT 模型的訓(xùn)練相似,其目標(biāo)是多次還原被掩碼的序列中的符號(hào)(氨基酸)。也就是說,基于已訓(xùn)練好的蛋白質(zhì)語言模型中的信息,以及當(dāng)前的蛋白質(zhì)結(jié)構(gòu)信息,對(duì)蛋白質(zhì)序列進(jìn)行多次改寫。LM-Design 基于全局序列信息對(duì)其中很少一部分符號(hào)(氨基酸)進(jìn)行改寫,所以對(duì)蛋白質(zhì)遠(yuǎn)距離依存關(guān)系能夠進(jìn)行很好的表示和預(yù)測。注:蛋白質(zhì)折疊之后,序列上距離很遠(yuǎn)的氨基酸在結(jié)構(gòu)上也可能很近。
現(xiàn)實(shí)中有大量經(jīng)過測序的蛋白質(zhì)序列數(shù)據(jù),但只有少量的蛋白質(zhì)結(jié)構(gòu)與序列對(duì)齊的數(shù)據(jù)。LM-Design 的一個(gè)優(yōu)勢是可以利用海量的蛋白質(zhì)序列數(shù)據(jù),充分學(xué)習(xí)和利用蛋白質(zhì)序列之間的進(jìn)化中產(chǎn)生的關(guān)聯(lián)關(guān)系,大幅提高從蛋白質(zhì)結(jié)構(gòu)到序列生成的預(yù)測準(zhǔn)確率。此外,我們發(fā)現(xiàn)增大預(yù)訓(xùn)練蛋白質(zhì)模型的規(guī)模可以進(jìn)一步提升準(zhǔn)確率。如圖 8 所示,LM-Design 是目前效果最好的蛋白質(zhì)序列生成模型,圖中圓的大小表示模型的參數(shù)量。
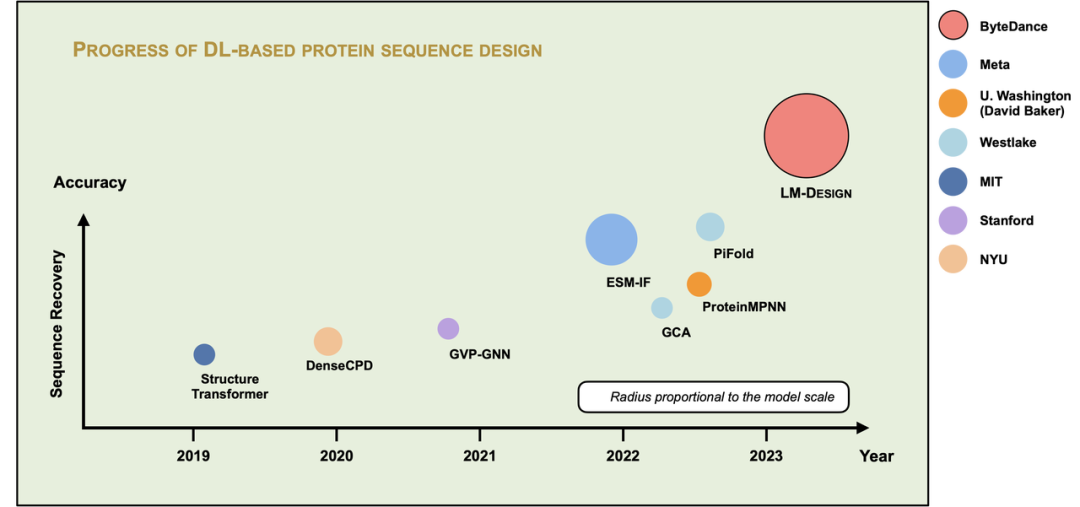
圖 8. 蛋白質(zhì)序列生成方法的精度
-
AI
+關(guān)注
關(guān)注
87文章
30137瀏覽量
268411 -
機(jī)器學(xué)習(xí)
+關(guān)注
關(guān)注
66文章
8377瀏覽量
132407 -
深度學(xué)習(xí)
+關(guān)注
關(guān)注
73文章
5492瀏覽量
120976
原文標(biāo)題:字節(jié)跳動(dòng)李航:AI for Science的一些探索和進(jìn)展
文章出處:【微信號(hào):zenRRan,微信公眾號(hào):深度學(xué)習(xí)自然語言處理】歡迎添加關(guān)注!文章轉(zhuǎn)載請(qǐng)注明出處。
發(fā)布評(píng)論請(qǐng)先 登錄
相關(guān)推薦
字節(jié)跳動(dòng)最新回應(yīng):正在探索AI芯片領(lǐng)域
《AI for Science:人工智能驅(qū)動(dòng)科學(xué)創(chuàng)新》第二章AI for Science的技術(shù)支撐學(xué)習(xí)心得
歸納AI領(lǐng)域一些方向的重要技術(shù)進(jìn)展





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論