 N雜五元芳烴的非共價成鍵機制研究
N雜五元芳烴的非共價成鍵機制研究
01引言
N雜五元芳烴在生物和藥物化學中占有重要地位,它是藥物分子的基礎結構單元,也是蛋白質、輔酶、生物堿、DNA等生物分子的核心骨架。由該基團參與的非共價相互作用(NCI)會介導分子識別、結合、催化和藥物傳遞等過程,從而在調節生物分子結構和諸多生命過程(如光合作用、信息存儲、DNA復制方面)中發揮著關鍵作用。因此,對N雜五元芳烴的非共價相互作用及其本質特征進行探究,有助于闡明許多復雜生物事件的物理化學機制。因此,本項目選取噻唑作為模型N雜五元芳香雜環化合物,采用高分辨轉動光譜結合理論計算方法精確解析其與四氟化碳、六氟化硫的非共價相互作用的拓撲幾何結構、結合強度和相互作用的本質。
02成果簡介
本項工作,基于密度泛函理論對噻唑-CF4和噻唑-SF6體系進行了幾何結構優化和振動頻率分析,進而在轉動光譜實驗中成功地指定和分配了屬于各自最穩定構象的轉動譜線,并結合量子化學理論計算闡明其分子間的非共價成鍵機制。實驗和理論計算的結果證實;所觀察到的噻唑-CF4異構體主要是由位于噻唑環平面的CF4的碳原子通過N···CCF4相互作用形成的,且每條轉動躍遷表現出由-CF3頂部內旋引起的A/E扭轉分裂;而噻唑和SF6主要通過范德華相互作用來穩定,SF6位于噻唑環上方。雖然靜電和色散都是噻唑-CF4和噻唑-SF6二聚體形成的主要因素,但在噻唑-SF6復合物中,色散項顯得尤其重要。另外,在比較幾種不同理論水平的計算精準度后發現,相較于M062X和PBE方法,B3LYP方法對噻唑-CF4復合物的計算更為準確,但對于噻唑-SF6復合物來說,ωB97XD方法得到的理論值的誤差更小。
03圖表導讀
(a)
(b)
(c)
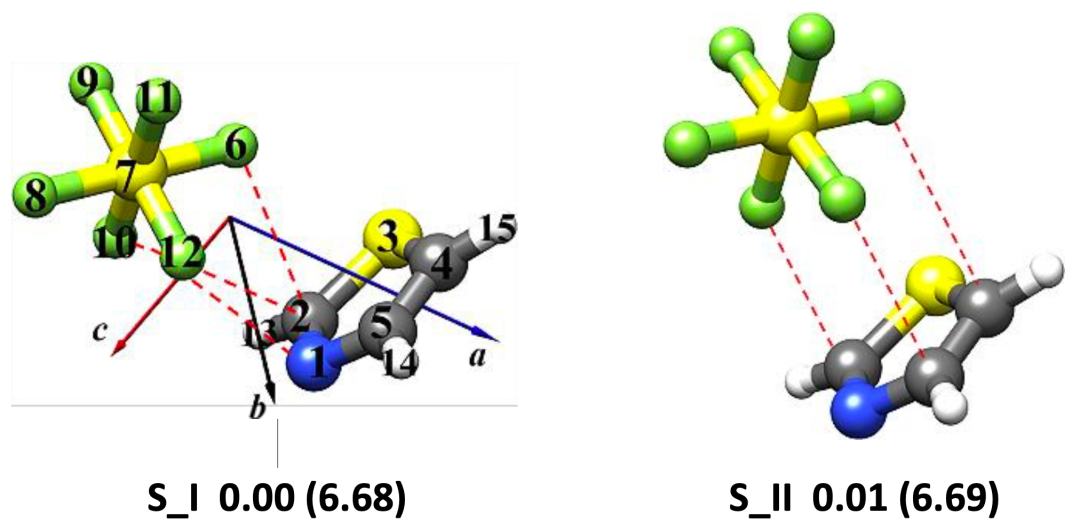
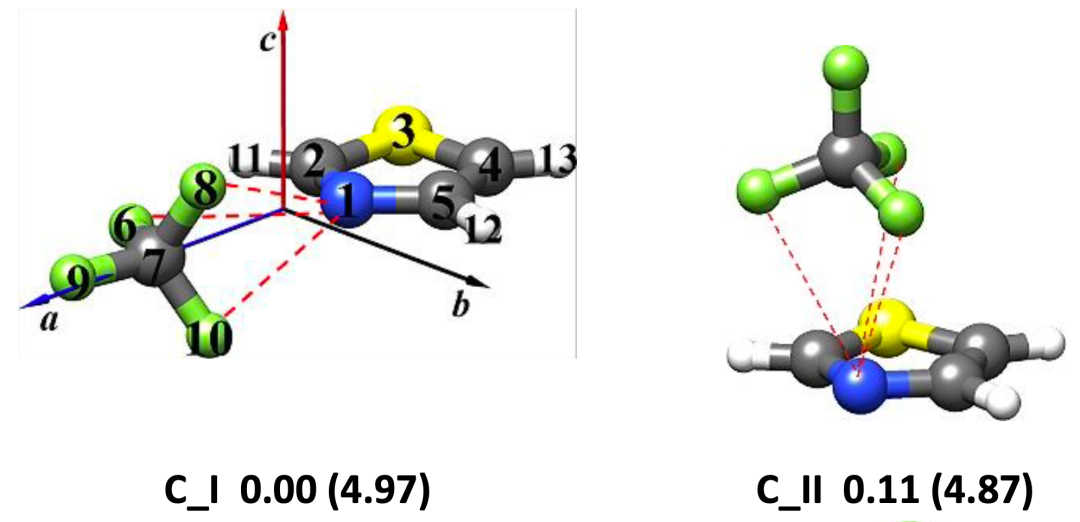
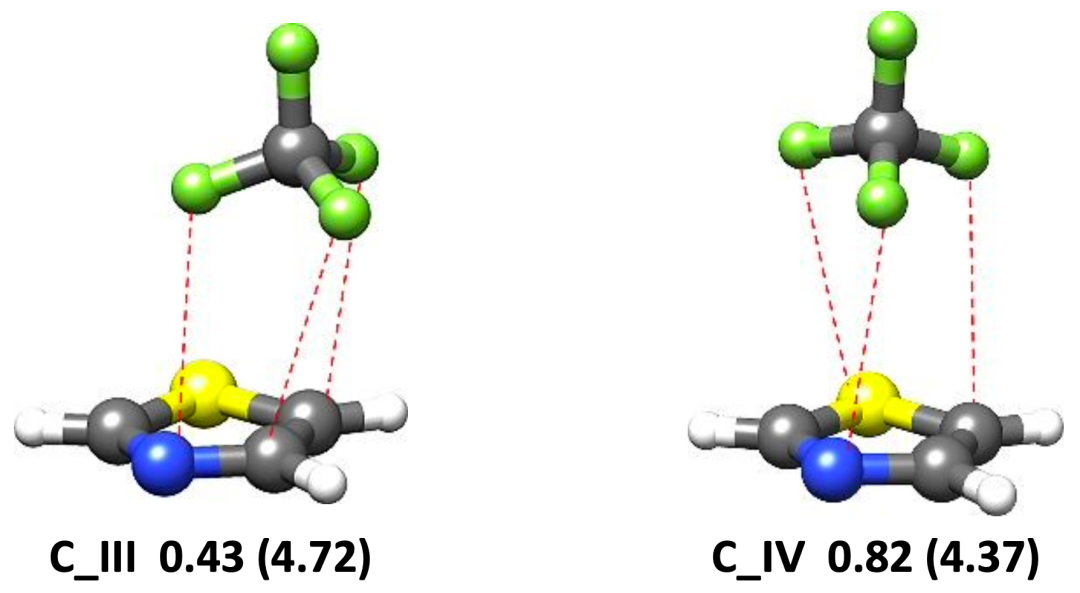
圖1.理論計算得到的噻唑-CF4(a)和噻唑-SF6(b)團簇的穩定構象
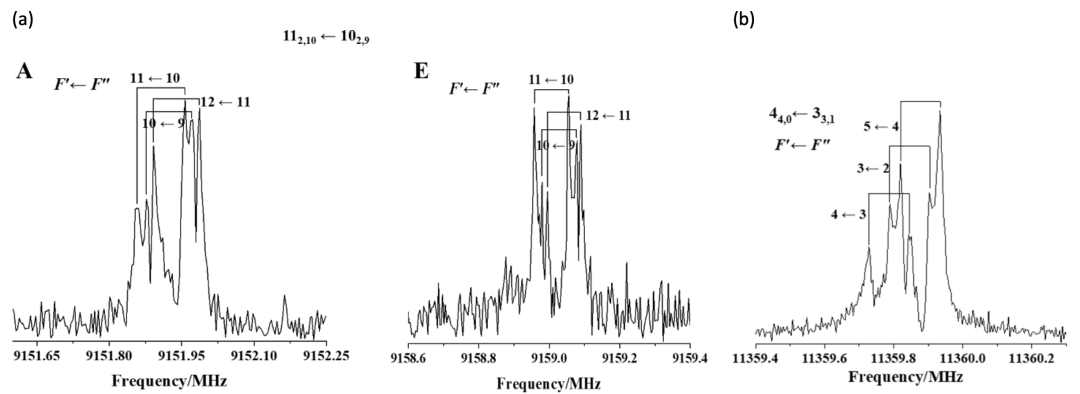
圖2.(a) thiazole-CF4的部分轉動光譜圖;(b) thiazole-SF6的部分轉動光譜圖
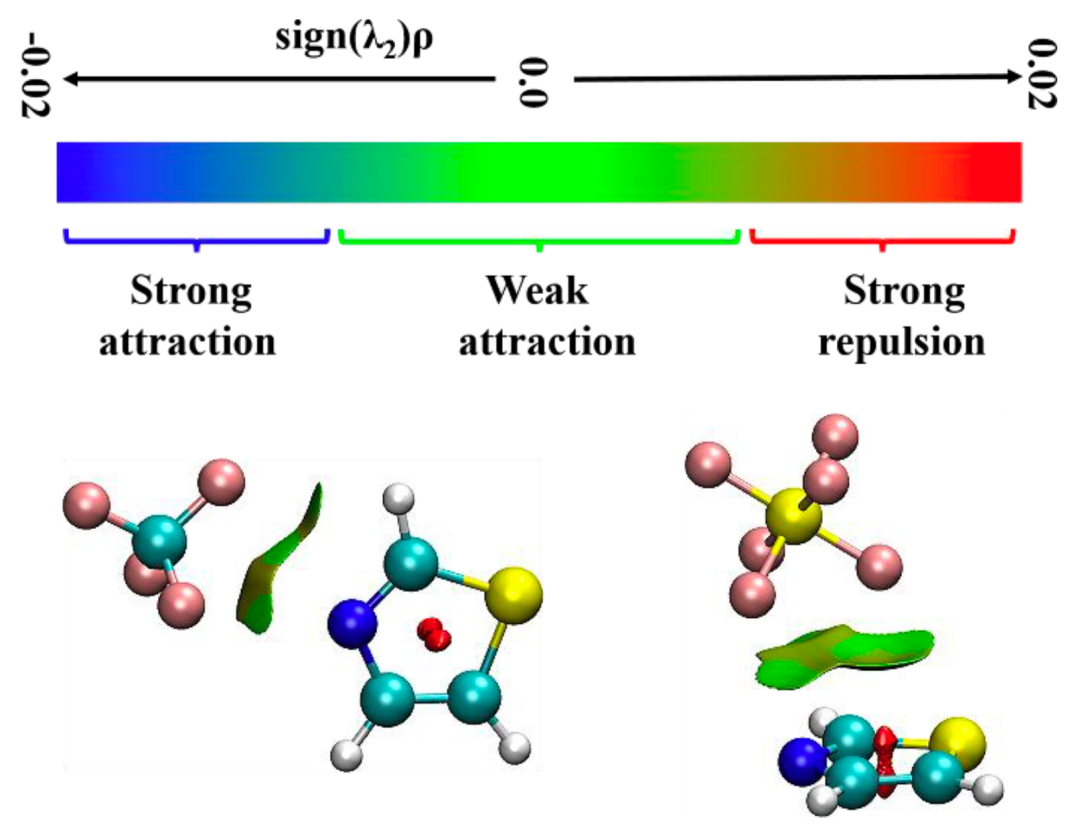
圖3.噻唑-CF4和噻唑-SF6團簇的簇內分子間非共價相互作用
表1.不同理論方法計算得到的thiazole-CF4的光譜參數
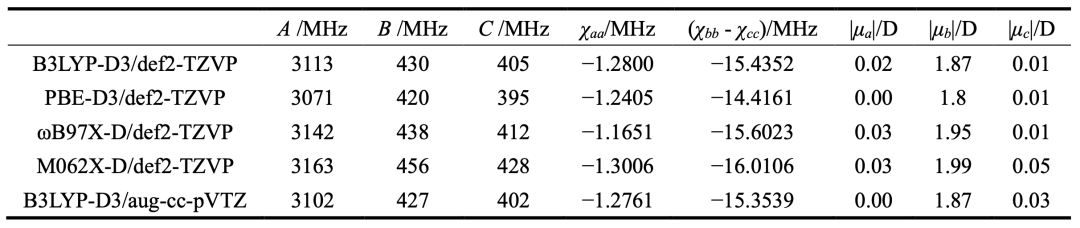
表2.不同理論方法計算得到的thiazole-SF6的光譜參數
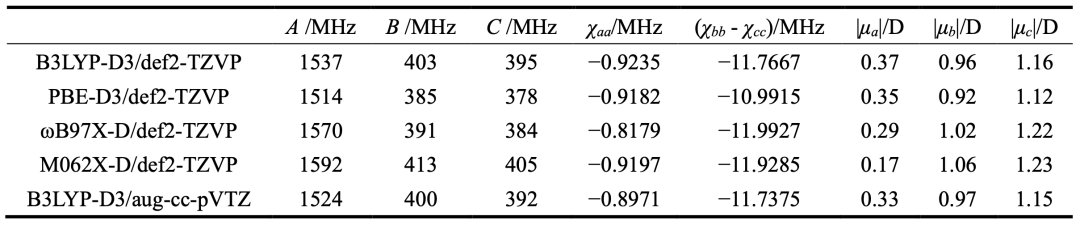
表3.實驗測定thiazole-CF4和thiazole-SF6的光譜參數

表4.thiazole-CF4和thiazole-SF6的SAPT分析的結果(kJ mol?1)
04小結
本文使用鴻之微Device Studio平臺搭載的BDF程序開展有關噻唑-CF4和噻唑-SF6體系的量子化學理論計算,再利用傅里葉變換微波光譜技術獲取各自最穩定構象的轉動光譜數據,從而確定分子結構并揭示非共價成鍵機制。研究表明,噻唑-CF4最穩定的構型是由靜電和色散主導的CF3···N相互作用拓撲,其rN···C距離為3.407 (2)A?;而位于噻唑環上方SF6是由范德華相互作用與噻唑結合,色散是主要吸引項。在理論計算方面發現,B3LYP-D3(BJ)/def2-TZVP理論水平能夠為噻唑-CF4和噻唑-SF6復合物提供與實驗相近的轉動常數值,且ωB97X-D3/def2-TZVP方法略微提高了噻唑-SF6的預測精度。
審核編輯:劉清
-
光譜儀
+關注
關注
2文章
947瀏覽量
30711 -
傅里葉變換
+關注
關注
6文章
437瀏覽量
42566
原文標題:文獻賞析 | N雜五元芳烴的非共價成鍵機制研究(馮剛)
文章出處:【微信號:hzwtech,微信公眾號:鴻之微】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
相關推薦
在中壓開關柜上使用微機五防鎖是否可以實現一鍵順控

鋁帶鍵合點根部損傷研究

模擬電子技術基礎電子版PDF第五版(童詩白,華成英)+答案詳解
金絲鍵合工藝溫度研究:揭秘鍵合質量的奧秘!

六類非屏蔽線纜和超五類非屏蔽線纜的區別
如何利用lighttools實現雜散光仿真呢?
引線鍵合在溫度循環下的鍵合強度衰減研究

鍵合銅絲的研究及應用現狀

光學設計中的雜散光
工藝參數對鍵合金絲質量影響的研究






 工商網監
工商網監
評論