") 固態(tài)電池中復(fù)合鋰陽極上固體電解質(zhì)界面的調(diào)控
固態(tài)電池中復(fù)合鋰陽極上固體電解質(zhì)界面的調(diào)控
研究背景
采用固體聚合物電解質(zhì)(SPE)的固態(tài)鋰金屬電池(SSLMB)具有更高的安全性和能量密度,在下一代儲能領(lǐng)域具有很大的應(yīng)用前景。隨著具有插層陽極的鋰離子電池(LIBs)的能量密度接近350wh kg?1的極限,金屬鋰(Li)成為SSLMB的一種極有前景的陽極,其理論比容量高達(dá)3860 mAh g?1。盡管存在這些優(yōu)勢,但在高面積容量(≥2.0 mAh cm - 2)時,還會產(chǎn)生大量“死”鋰和不穩(wěn)定SEI等挑戰(zhàn),阻礙了電池的進(jìn)一步發(fā)展。雖然已經(jīng)提出了界面工程、電解質(zhì)改性和使用復(fù)合鋰陽極等方法來克服這些障礙并延長電池的壽命,但在過去的幾十年里,SPE和Li陽極之間SEI的演變和作用通常被忽視,對不同鋰陽極SEI的結(jié)構(gòu)和組成聚合物基固態(tài)電解質(zhì)的性能仍然知之甚少。SEI結(jié)構(gòu)決定了運輸速率以及從電解質(zhì)到陽極的Li離子(Li+)的均勻性,顯著影響速率和極化陽極/電解質(zhì)界面上的電化學(xué)反應(yīng)。所以了解復(fù)合陽極對SEI中各種化合物形成的影響,對于掌握陽極對鋰沉積動力學(xué)的影響至關(guān)重要。又由于在循環(huán)過程中,持續(xù)的SEI降解和再生會導(dǎo)致容量衰退,因此鋰金屬/SPE界面上的固體電解質(zhì)界面(SEI)的穩(wěn)定性是一個主要挑戰(zhàn)。
成果簡介
近日,北理工前沿交叉院黃佳琦教授聯(lián)合清華大學(xué)張強教授共同研究了固態(tài)鋰金屬電池中鋰陽極(l-SEI)和復(fù)合鋰陽極(c-SEI)上SEI的形成對整體電化學(xué)性能的影響。通過研究發(fā)現(xiàn),復(fù)合陽極會形成均勻的富含Li2S的無機SEI層和較薄的有機SEI層,能夠有效地鈍化界面以增強循環(huán)穩(wěn)定性。并且,在2. 0mAhcm-2的高面容量下,帶有c-SEI鋰陽極的全電池在0. 5C下可維持400多次循環(huán)。此外,即使在卷曲和切割后,可逆的高陽載固態(tài)軟包電池也表現(xiàn)出卓越的安全性。這些發(fā)現(xiàn)為開發(fā)具有強大SEI的復(fù)合電極為基于固態(tài)聚合物的鋰金屬電池提供了寶貴的見解。此工作以”The Regulation of Solid Electrolyte Interphase on Composite LithiumAnodes in Solid-State Batteries“為題,發(fā)表在Angewandte Chemie上。
研究亮點
1.提出了一種利用富Li2S結(jié)構(gòu)實現(xiàn)的復(fù)合陽極電極的表面優(yōu)勢形成致密鈍化SEI的策略。
2.采用三電極技術(shù)研究了c-SEI在界面上的鈍化效應(yīng)如何有效抑制聚醚的寄生界面反應(yīng)。
3.具有LiFePO4(LFP)陰極和c-SEI保護陽極的全電池在0.5℃下保持400多次循環(huán),面積容量為2.0mAh cm?2。
4.通過合理設(shè)計SSLMB中的復(fù)合陽極電極可誘導(dǎo)SEI界面的非均勻化,為實現(xiàn)高性能SSLMB提供了機會。
圖文導(dǎo)讀
在這項工作中,作者提出了一種利用復(fù)合陽極電極表面優(yōu)勢形成致密鈍化SEI的策略,探討了SSLMB中金屬Li陽極和復(fù)合Li陽極上SEI的演變。復(fù)合陽極上的SEI(c-SEI)是通過富Li2S結(jié)構(gòu)實現(xiàn)的。

圖1所示。Li金屬和復(fù)合Li陽極上形成的l-SEI和c-SEI微觀結(jié)構(gòu)。(a)和(d)低溫透射電鏡圖像描繪了鋰金屬和(d)復(fù)合鋰陽極的形貌。(b) 低溫透射電鏡圖像顯示l-SEI和(e)c-SEI具有明顯的內(nèi)有機層和外非晶態(tài)層。(c) l-SEI和(f)c-SEI的SAED圖像提供了對組件的進(jìn)一步了解。
為了直接探測SEI的微觀結(jié)構(gòu),通過低溫透射電子顯微鏡(cryo-TEM)對純鋰陽極(l-SEI)和復(fù)合陽極(cSEI)上的鋰金屬SEI層進(jìn)行了表征。提出了一種雙銅(Cu)網(wǎng)結(jié)構(gòu),用于直接觀察固態(tài)電池中鋰陽極上的鋰和SEI,而不是常規(guī)的溶劑分散或電化學(xué)沉積方法在1.0 mA cm?2的恒定電流密度下鍍Li 1.0 h。圖1a–c顯示了Cu上Li和l-SEI的代表性圖像,其中Cu表面出現(xiàn)了明顯的Li枝晶生長。
l-SEI的內(nèi)層含有較少的無機化合物,如Li2O,而外層比cSEI厚(圖1b)。CP表面均勻的Li金屬鍍層(圖1d)有利于可逆沉積和剝離。低溫TEM圖像顯示了一種典型的雙層結(jié)構(gòu),其中c-SEI包括一個15 nm厚的非晶層和一個富含Li2S、Li2O和LiF的無機層(圖1e)。c-SEI中的有機層厚度減少了約20%,這有助于減少均勻鋰離子通量的擴散路徑長度。相應(yīng)的選區(qū)電子衍射(SAED)證實了這些無機晶體材料的存在,并檢測到Li2CO3的衍射環(huán)和幾個單晶斑點圖案(圖1f)。c-SEI含有Li2CO3納米晶體。此外,單晶斑點圖案的存在表明存在LiCx結(jié)構(gòu),這可以增強c-SEI的親鋰性能。c-SEI層的SAED顯示出LiF、Li2O、Li2CO3和Li2S的顯著存在,而l-SEI缺乏與Li2S相對應(yīng)的衍射環(huán)(圖1c和1f)。上述結(jié)果表明,當(dāng)沉積鋰金屬時,初始基板可以決定后續(xù)的SEI。
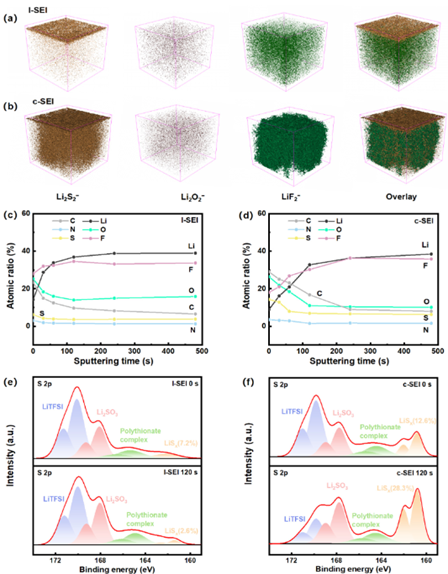
圖2:通過ToF-SIMS和XPS分析揭示了l-SEI和c-SEI的3D納米結(jié)構(gòu)。Li2S2-(代表Li2S)、Li2O2-(代表LiO)、LiF2-(代表LiFe)及其在(a)l-SEI和(b)c-SEI的ToF-SIMS濺射體積內(nèi)的疊加的3D視圖。此外,通過XPS測試,在(c)l-SEI和(d)c-SEI上形成的SEI內(nèi)元素的分布。XPS光譜進(jìn)一步證明了S 2p在(e)l-SEI和(f)c-SEI上v的分布。
單個無機組分的三維結(jié)構(gòu)和空間分布l-SEI和c-SEI上的組件分別如圖2a和2b所示。Li2S2-含量在最初的50秒內(nèi)迅速降低,隨后強度接近0。這一觀察表明,Li2S在內(nèi)部l-SEI中的存在最小。相比之下,在CP上形成的SEI要薄得多,包括外層致密的有機物質(zhì)層和富含Li2S的無機層。由復(fù)合陽極形成的無機層阻礙了pDOL的持續(xù)分解,從而減小了有機層的厚度。由于FEC的自有損耗,LiF在兩個 andes 上的分布和梯度相似。
為了表征不同陽極的SEI結(jié)構(gòu),采用X射線光電子能譜(XPS)來鑒定無機和有機成分。根據(jù)不同濺射時間的元素分布,SEI的成分在最初的60秒內(nèi)發(fā)生變化,并在120秒后穩(wěn)定下來(圖2c和2d)。因此,選擇0秒和120秒的XPS數(shù)據(jù)來比較SEI內(nèi)層和外層的組成和含量。此外,含硫物種的增加也表明了復(fù)合陽極對電解質(zhì)分解的影響。圖2e和2f分別顯示了l-SEI和c-SEI上的去卷積S 2p光譜。具體來說,在169.4/170.2、167.3/168.5、161.4/160.2和158.3/157.1處出現(xiàn)了四個不同的S 2p雙峰,分別對應(yīng)于S=O、S-O-Li、多面體絡(luò)合物和Li-S。c-SEI顯示S=O的存在減少,表明TFSI?分解程度增加。此外,結(jié)構(gòu)內(nèi)可辨別的Li2S豐度(內(nèi)層28.3%)與cryoTEM和ToF-SIMS的早期發(fā)現(xiàn)一致,表明c-SEI內(nèi)有Li2S富集的傾向。
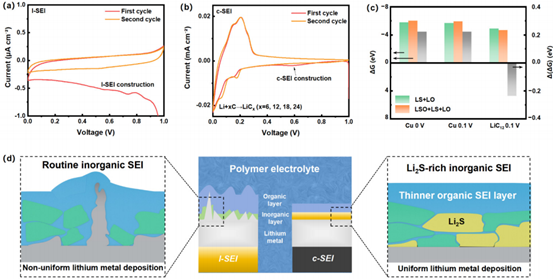
圖3:富li2s無機SEI形成的成因。(a) l-SEI和(b) c-SEI的初始和第二循環(huán)伏安(CV)圖像。隨附自由能計算結(jié)果,ΔGm表示涉及TFSI -還原到其各自產(chǎn)物的單電子反應(yīng)的自由能變化。通過ΔGm2減去ΔGm1得到Δ(ΔGm)。(d)常規(guī)l-SEI和c-SEI的SEI示意圖,其中c-SEI表現(xiàn)為均勻的富含li2s的無機SEI層和較薄的有機SEI層。
圖3a和3b顯示了l-SEI和c-SEI從0到1 V的循環(huán)伏安法(CV)曲線。在初始循環(huán)中,CP觀察到LiTFSI的明顯降解過程,初始陰極峰為0.6 V。隨后,第二個循環(huán)中0.6 V陰極峰的消失表明形成了穩(wěn)定的SEI。此外,0.18和0.07 V的陰極峰表明CP中的Li嵌入過程,分別對應(yīng)于LiC12和LiC6的形成。因此,與銅相比,CP的使用提高了TFSI-陰離子在大電流下的還原程度,為c-SEI中硫含量較高提供了合理的解釋。
為了進(jìn)一步闡明陽極如何影響反應(yīng)硫化物化合物,我們計算了這兩個反應(yīng)的吉布斯自由能變化(ΔG)(圖3c)。該計算可作為評估反應(yīng)趨勢的熱力學(xué)描述符。在這種設(shè)置中,Cu電勢設(shè)置為0或0.1 V,鋰化石墨電極電勢固定為0.1V。計算出的ΔG值由電子數(shù)標(biāo)準(zhǔn)化,從而得出TFSI?還原反應(yīng)的每電子轉(zhuǎn)移ΔG(ΔGm)。
為了比較不同陽極上兩種反應(yīng)的趨勢,我們計算了ΔGm的差值(Δ(ΔGm)=ΔGm,eq1-ΔGm、eq2)。在使用相同電極的0和0.1 V下,Li2S2O4表現(xiàn)出相同的反應(yīng)模式(Δ(ΔGm)均為0.22 eV),表明對反應(yīng)趨勢的潛在影響很小。然而,在相同電勢下,Li2S2O4在不同陽極上表現(xiàn)出不同的趨勢,LiC12的Δ(ΔGm)為-0.23eV,而Cu的Δ為0.22eV。這表明當(dāng)表面從l-SEI變?yōu)閏-SEI時,Li2S2O3更傾向于還原為無機Li2S。上述結(jié)果表明,陽極類型與SEI演變之間存在顯著相關(guān)性。CP的表面傾向于分解TFSI?陰離子,從而產(chǎn)生富含Li2S的鈍化層。綜上所述,TFSI-陰離子和c-SEI中主要無機物種Li2S的分解有助于構(gòu)建穩(wěn)定的無機SEI層。
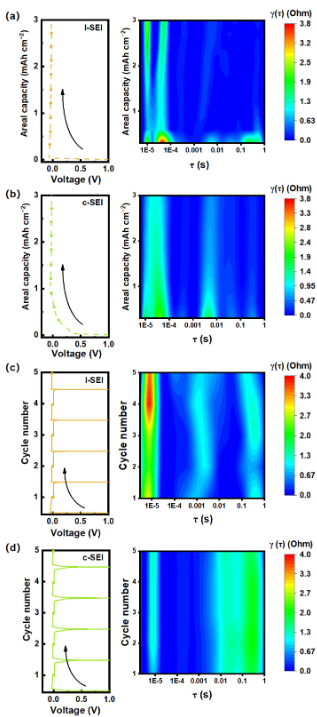
圖4:鍍鋰和剝離過程中SSLMB三電極EIS評價的DRT轉(zhuǎn)變分析。圖(a)和圖(b)分別為鋰陽極和復(fù)合陽極的初始沉積和阻抗時間分布。并分別給出了(c)鋰陽極和(d)復(fù)合鋰陽極5次循環(huán)的電壓周期曲線和DRT分析。所有循環(huán)均在3.0 mAh cm - 2和1.0 mA cm - 2下執(zhí)行。
為了準(zhǔn)確監(jiān)測鋰沉積動力學(xué)的變化,在內(nèi)置聚合物體系中的三電極配置中,記錄了初始周期(圖4a和4b)和擴展周期(圖4c和4d)的阻抗譜。采用三電極結(jié)構(gòu),以Li@Cu為參考電極,用于精確測量陽極的阻抗信號,有效地避免了Li對電極產(chǎn)生的問題。
時間尺度識別是將非破壞性阻抗特性與電池實時監(jiān)測相結(jié)合的關(guān)鍵方法。利用三電極系統(tǒng)和電化學(xué)阻抗譜(EIS)數(shù)據(jù)的弛豫時間分布(DRT)分析,跟蹤了界面阻抗的演變。所得的DRT圖允許通過陽極上不同的弛豫時間來識別特定的電化學(xué)過程。在10?5到10?4 s的τ范圍內(nèi),阻抗明顯為峰值,表示SEI響應(yīng)。此外,在0.001 ~ 0.01 s之間的τ反映了電荷轉(zhuǎn)移電阻,而0.1 s以下的τ反映了Li離子的擴散。在初始電鍍周期中,l-SEI上的RSEI初始值為3.8 Ω,大于c-SEI的1.5 Ω,并且占據(jù)了阻抗的主要部分(53.3%),表明SEI生長不均勻(圖4a)。
因此,不均勻的鍍剝離機制在整個循環(huán)過程中產(chǎn)生額外的SEI,導(dǎo)致RSEI持續(xù)增加。然而,c-SEI在第一次沉積過程的整個時間尺度識別中表現(xiàn)出穩(wěn)定的RSEI (1.5 Ω),具有較高的鋰剝離能力(圖4b)。Li金屬可以保持穩(wěn)定的生長,避免了由于SEI不均勻而導(dǎo)致的界面Li+通量不均勻。在整個循環(huán)過程中,與c-SEI的穩(wěn)定狀態(tài)相比,l-SEI上的RSEI明顯增加(圖4d和4e)。這表明Li+在通過c-SEI時遇到的阻礙較小。
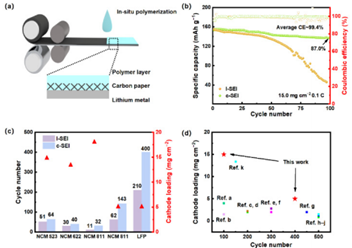
圖5:l-SEI和c-SEI陽極的電化學(xué)性能。(a)利用軋制法制造c-SEI陽極的工藝示意圖。(b) 15.0 mg cm?2 LFP電極的長期循環(huán)性能。(c)不同陰極和相應(yīng)面陽荷下的長期循環(huán)性能。(d) SSLMB陽極材料載荷和循環(huán)次數(shù)的比較。
在室溫下,將超薄鋰箔直接軋制到碳紙主體/平面銅箔上,形成復(fù)合鋰陽極(圖5a)。
隨后的原位聚合工藝用于制造集成復(fù)合陽極,有利于大規(guī)模生產(chǎn)。電壓的升高表明,在電鍍過程中引發(fā)鋰成核的難度加大,而在循環(huán)的反轉(zhuǎn)階段引發(fā)剝離的難度加大。這種現(xiàn)象通常與電極處大量SEI的發(fā)展和工作界面處電子隔離或死鋰的積累有關(guān)。當(dāng)死鋰和具有電子絕緣的SEI的積累達(dá)到臨界點時,極化急劇增加,最終導(dǎo)致350 h后鋰鋰對稱電池失效。這些發(fā)現(xiàn)表明,c-SEI有效地抑制了鋰與電解質(zhì)之間的不良反應(yīng),確保了在重復(fù)鍍鋰/剝離過程中保持高可逆性。
當(dāng)溫度恢復(fù)到0.2℃時,LFP| C-SEI陽極保留了98.0%的初始容量,表現(xiàn)出顯著的穩(wěn)定性。相比之下,LFP|l-SEI陽極的容量保持率明顯下降至85.4%左右,這表明Li陽極在充放電過程中存在固有的不穩(wěn)定性和較差的可逆性。當(dāng)c-SEI陽極與高陽載陰極(15.0 mg cm?2)配對時,充滿的電池仍然可以保持高容量和長壽命。具有裸金屬鋰的高陽載LFP全電池在60個循環(huán)中表現(xiàn)出快速的容量衰減(圖5b),這歸因于高容量下緩慢的鋰沉積動力學(xué),導(dǎo)致死鋰和SEI積累增加。然而,高陽載LFP|c-SEI陽極電池表現(xiàn)出令人印象深刻的容量保持,保持了其初始放電容量的87.0%(132.1 mAh g?1)。構(gòu)建和穩(wěn)定一致的Li+輸運路徑的結(jié)合增強了Li+離子通量的均勻分布。
當(dāng)與不同類型和陽載的NCM陰極匹配時,c-SEI陽極與l-SEI陽極相比,在循環(huán)壽命方面取得了令人印象深刻的改善(圖5c)。快速的容量衰落歸因于pDOL在高壓下的固有不穩(wěn)定性,但它仍然突出了推進(jìn)高能量密度SSLMB的潛力。c-SEI陽極在SSLMB中表現(xiàn)出優(yōu)異的電化學(xué)性能,與SSLMB相關(guān)的陰極材料陽載和當(dāng)前作品的循環(huán)次數(shù)的比較如圖5d所示。如上所示,我們在面積陽載和循環(huán)壽命方面取得了更顯著的進(jìn)展,這可以歸因于復(fù)合鋰陽極上固體電解質(zhì)界面相的調(diào)節(jié)。
總結(jié)與展望
本文作者探討了SSLMB中金屬Li陽極和復(fù)合Li陽極上SEI的演變,提出了一種利用復(fù)合陽極電極表面優(yōu)勢形成致密鈍化SEI的策略。具體來說,復(fù)合電極表面促進(jìn)了LiTFSI分解成富含li2s的無機SEI層,pDOL分解成薄的有機SEI層。采用這種無添加劑策略,我們成功地獲得了富含li2s的SEI層,作為陽極/SPE表面的鈍化層。在高面積容量(3.0 mAh cm?2)下,庫侖效率(>98.5%)得到提高,循環(huán)壽命延長。特別是通過簡單的一步法制備的復(fù)合陽極,在實際應(yīng)用中得到了有效的應(yīng)用。完整的電池還表現(xiàn)出令人印象深刻的長期循環(huán)穩(wěn)定性,并在0.5 c下超過400次循環(huán)保持穩(wěn)定的122.5mAh g- 1比容量。此外,復(fù)合陽極與超過15.0 mg cm - 2的高陽載陰極配對時,在固態(tài)袋狀電池中表現(xiàn)出優(yōu)異的可逆性和安全性。在各種條件下。這一貢獻(xiàn)為SEI的結(jié)構(gòu)演變提供了有價值的見解,強調(diào)了陽極材料對工作SSLMB中SEI形成的關(guān)鍵影響。
-
鋰離子電池
+關(guān)注
關(guān)注
85文章
3215瀏覽量
77562 -
電解質(zhì)
+關(guān)注
關(guān)注
6文章
805瀏覽量
20019 -
固態(tài)電池
+關(guān)注
關(guān)注
9文章
692瀏覽量
27702
原文標(biāo)題:北理工黃佳琦教授聯(lián)合清華張強教授Angew.Chem:固態(tài)電池中復(fù)合鋰陽極上固體電解質(zhì)界面的調(diào)控
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關(guān)注!文章轉(zhuǎn)載請注明出處。
發(fā)布評論請先 登錄
相關(guān)推薦
新型固體材料可替代電池中的易燃液體電解質(zhì)
剖析穩(wěn)定鋰金屬電池的長效固體電解質(zhì)界面
固態(tài)鋰金屬電池中的電解質(zhì)-負(fù)極界面保護層
氟化石墨烯增強聚合物電解質(zhì)用于固態(tài)鋰金屬電池
鈉離子電池的電解質(zhì)分類
改變電解質(zhì)分布調(diào)控固態(tài)界面實現(xiàn)高性能固態(tài)電池
固態(tài)電解質(zhì)引入特殊官能團實現(xiàn)高電壓鋰金屬固態(tài)電池
如何有效構(gòu)建固體電解質(zhì)的高親鋰界面?
固態(tài)電池電解質(zhì)的分類及性能對比
鋰金屬電池的鋰微觀結(jié)構(gòu)與固體電解質(zhì)界面之間的關(guān)系
聚合物電解質(zhì)離子電導(dǎo)率及界面穩(wěn)定性的影響因素
固態(tài)電池中鋰枝晶的起源與調(diào)控
認(rèn)識石榴石固態(tài)電解質(zhì)的表面再生和反應(yīng)性
鈮酸鋰調(diào)控固態(tài)電解質(zhì)電場結(jié)構(gòu)促進(jìn)鋰離子高效傳輸!
通過電荷分離型共價有機框架實現(xiàn)對鋰金屬電池固態(tài)電解質(zhì)界面的精準(zhǔn)調(diào)控





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評論