 電子發燒友App
電子發燒友App


高熵水系電解質全氣候鋅電池
【背景】
交通工具的電氣化和電網儲能需求的上升,繼續在全球范圍內形成圍繞電池的勢頭。然而,鋰離子電池的供應鏈面臨著基本和稀缺材料資源的日益挑戰。因此,開發更多可持續的電池化學制品的動機正在增長。
【工作介紹】
本工作展示了一種引入氯化鋰作為支撐鹽的氯化鋅水溶液電解質。采用優化的電解質Li2ZnCl4?9H2O,組裝的鋅空氣電池可以在-60℃和+80℃之間以0.4 mAcm-2的電流密度維持800小時的穩定循環,鋅剝離/電鍍的庫倫效率為100%。即使在-60℃,也可以保留>80%的室溫功率密度。
通過先進的表征和理論計算表明:高熵的溶劑化結構是其卓越性能的原因。強酸性使ZnCl2接受提供的Cl-離子,形成ZnCl4?2-陰離子,而水分子在低鹽濃度下保持在自由溶劑網絡內或與Li離子配位。
Li2ZnCl4·9H2O電解質中,Li?Cl解離、[ZnC4?m2?m]n聚集體長度的減少和游離溶劑氫鍵網絡的破壞之間的競爭產生了具有高熵的獨特的溶劑化結構,有助于保持優異的離子電導率,其中溶劑得到了極大的穩定,結晶受到抑制。
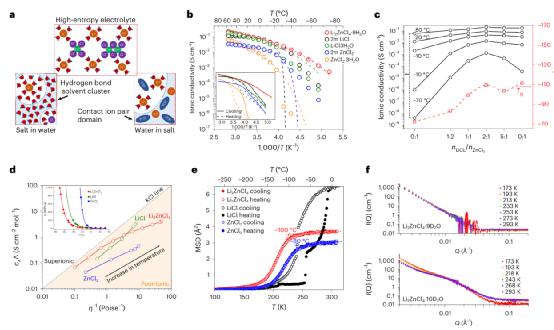
圖1、寬溫窗下的傳輸特性。
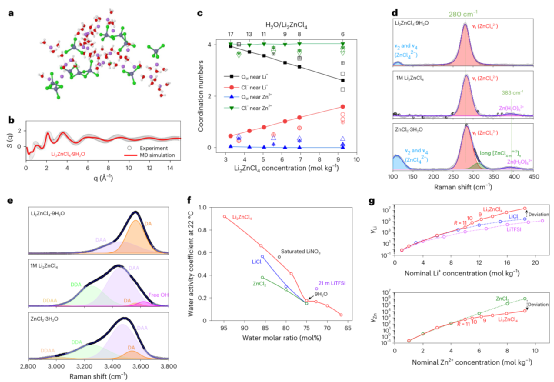
圖2、溶劑化結構。
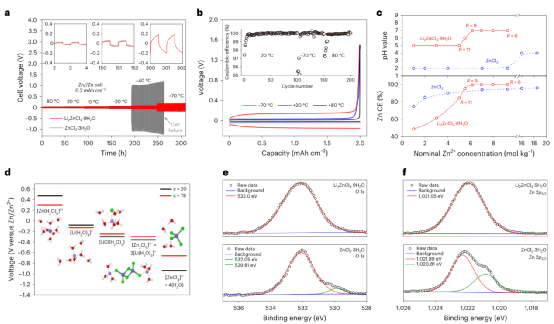
圖3、鋅金屬陽極性能。
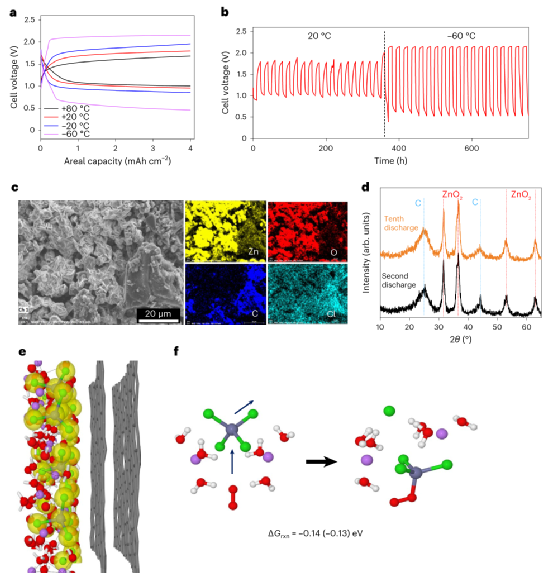
圖4、鋅-空氣電池性能。
利用該電解質,同時實現了廣泛的電化學穩定性窗口(優于鹽水電解質)和特殊的操作溫度范圍(-80℃至+80℃,僅在該電解質中發現)。研究工作證明了該電解質的溶劑化結構、傳輸和電化學穩定性之間的關系,提供了一個理解的基礎,激發了未來水基金屬離子電池的電解質設計。
作為高熵溶劑化環境概念功效的證明,在-60℃至20℃的溫度范圍內,鋅-空氣電池實現了前所未有的穩定性。盡管含氯電解質在商業原電池(如鋅-二氧化錳電池、鋅-碳電池、鋰-亞硫酰氯電池)中得到廣泛使用,并被提議用于可充電的多價離子電池(如鋅-離子、鎂-離子、鋁-離子),但在未來的應用中可能需要通過防腐涂層、抑制劑或碳化集流體來解決腐蝕問題。
用于高性能鋰硫電池的高負載Li2S陰極中的長期加強電子網絡
【背景】
實現具有高能量密度和長壽命的硫化鋰(Li2S)正極需要創新的正極設計,以最大限度地提高電化學性能和抵抗電極退化。
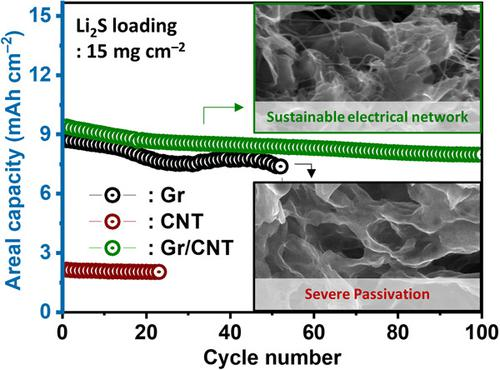
【工作介紹】
本工作開發了一種基于高負載Li2S的陰極,該陰極具有由二維石墨烯(Gr)和一維碳納米管(CNTs)組成的緊湊幾何形狀的微米級Li2S顆粒,并強調了CNTs在高容量Li-S電池穩定循環中的作用。在一個維度組合的碳基體中,嵌入Gr片內的CNTs創造了強大和可持續的電子擴散途徑,同時抑制了活性碳表面的鈍化。
作為一個獨特的點,在第一次充電過程中,所提出的陰極通過Li2S直接轉化為S8?,而不誘發多硫化鋰的形成而被完全激活。
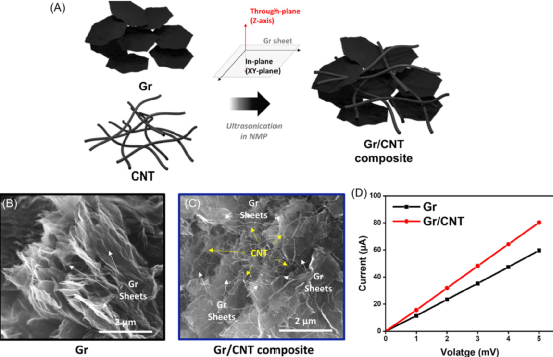
圖 石墨烯(Gr)/碳納米管(CNT)(Gr/CNT)復合材料的特征。
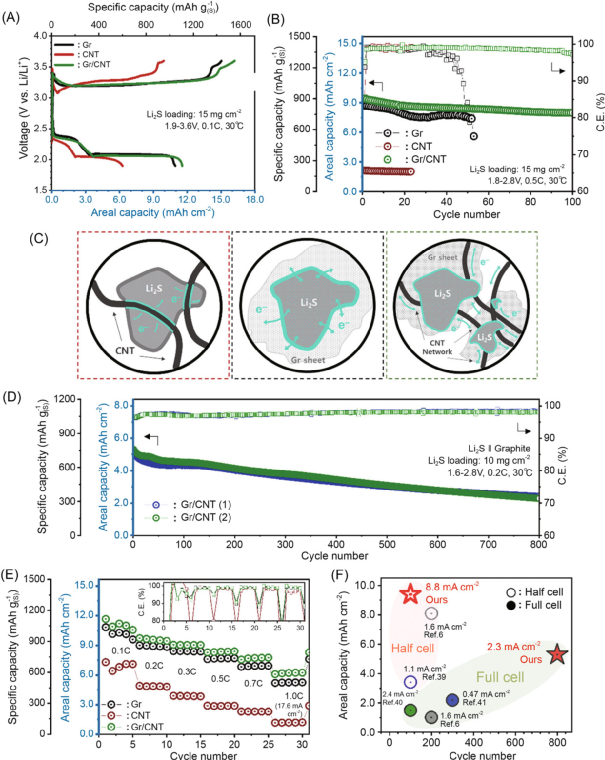
圖 緊湊型Li2S/Gr/CNT陰極在鋰-S電池中的電化學性能。
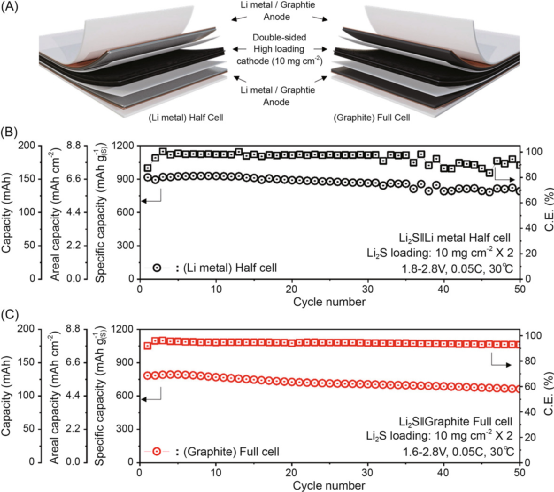
圖 使用緊湊的Li2S/Gr/CNT陰極制造的軟包半電池和全電池。
【結論】
利用高壓造粒法提出了一種合理設計的緊湊型Li2S/Gr/CNT陰極。Gr的平面形態允許與散裝Li2S的大面積接觸,從而誘導Li2S的簡易活化,而CNT形成的電網絡在長時間的循環中保持了均勻、簡易的反應。此外,陰極的緊湊幾何形狀使碳基體和活性材料之間能夠緊密接觸。與以前報道的基于Li2S的鋰硫電池的結果相比,使用緊湊型Li2S/Gr/CNT陰極的半電池和全電池顯示出前所未有的高面積容量和循環壽命。
值得注意的是,在緊湊型Li2S/Gr/CNT陰極中觀察到了第一次充電時的直接轉換反應(從Li2S到S8?),并使用原位技術和低溫TEM進行了研究。在緊湊型Li2S/Gr/CNT陰極中,高導電性的碳基體、電極的緊湊幾何形狀、高度變形的Li2S(在高壓造粒過程中形成)以及這種獨特的直接轉換反應都有助于有效地激活Li2S。
值得注意的是,即使在低電解質條件下(電解質量與Li2S的重量比為7μL mg–1?),陰極也顯示出很高的電化學活性,在0.2C下的實際面積容量為~9.7 mAhcm–2?。最后,還展示了基于大尺寸軟包Li2S的半電池和Li-S全電池,其Li2S的高負載為10mg cm–2?,為Li-S電池中陽極選擇的重要性提供了見解。
基于Li2S的Li-S電池的優勢,可以避免與Li金屬陽極的安全隱患有關的問題,將有效地加速Li-S電池的實際使用。這種新提出的陰極以及本研究中觀察到的電化學性能將為鋰硫電池的進一步發展提供有益的指導。
提高石榴石固體電解質離子電導率的非平衡動力學
【背景】
固體電解質(SSE),作為所有固態電池的重要組成部分,仍然面臨著離子導電性有限的關鍵困境。通過摻雜雜價過渡金屬來調節Li+?離子的部分占有率,是實現高Li+?電導率的一個重要特征。然而,其結構和動力學機制仍不清楚,妨礙了合理設計更高的電導率的SSE。
【工作介紹】
本工作以Li7-xLa3Zr2-xTaxO12?(0≤x≤0.625)的典型石榴石SSE為例,發現Ta5+?-摻雜濃度x=0.25導致大量的非平衡Li+?構型為[LiO6?]-[LiO4?]-[VLiO6] 。這種非平衡構型執行高偏移和高靜電能量的Li+?離子,減少了Li+?-離子傳輸的激活能。
因此,雜價離子的摻入通過控制非平衡Li+?離子的數量,對Li+?- 離子導電性有很大的影響。這些發現為理解離子傳輸提供了重要的啟示,并為優化Li+?的分布以提高離子導電性鋪平了道路。
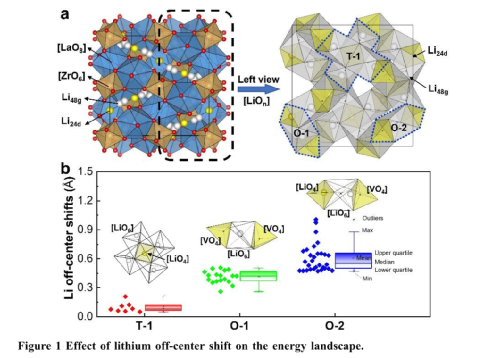
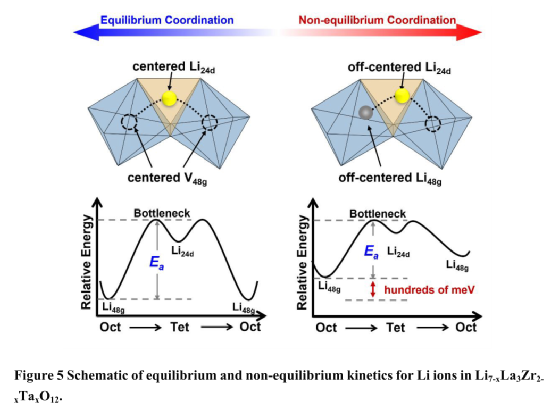
作者系統地探討了Li7-xLa3Zr2-xTaxO12中Li+離子的非平衡動力學對離子傳導性的貢獻。基于DFT和AIMD計算發現:非平衡構型中的Li24d離子具有較高的偏移和較高的靜電能量以加速Li+離子的擴散,而平衡和非平衡構型中的Li48g離子顯示出比較的偏移和靜電能量。
在[Li48gO6]-[Li24dO4]-[V48gO6]的T-2構型中,Li+離子的非平衡動力學來源于雜價Ta5+離子的取代和Li48g位點的空位的引入。對Ta5+摻雜濃度影響的研究表明,隨著Ta5+濃度的增加,非平衡Li24d離子的數量呈現先增加后減少的趨勢。Li6.75La3Zr1.75Ta0.25O12(Li7-xLa3Zr2-xTaxO12中的x=0.25)具有最高數量的非平衡T-2構型,并獲得最低的活化能。
這些發現為理解原子層面的離子傳輸提供了重要的見解,并有助于設計和優化下一代無機SSE。
為全固態電池設計嵌入型無鋰陰極的定制策略
【背景】
將無鋰過渡金屬陰極(MX)與鋰金屬陽極配對,是克服當前可充電鋰離子技術的能量密度限制的一個新興趨勢。然而,由于長期被忽視的電壓調整/相位穩定性競爭,實用的無鋰MX陰極的開發受到現有的低電壓概念的困擾。
【工作介紹】
本工作提出了一種涉及三個電壓/相位演化階段的p型合金化策略,其中每個階段的變化趨勢都由兩個改進的配位體-場描述符來定量,以平衡上述矛盾。在此之后,成功地設計了一種從層狀MX2?材料中的插層型2H-V1.75Cr0.25S4?陰極,它在電極水平上擁有554.3 Wh kg?1?的能量密度,同時與硫化物固態電解質的界面兼容。
這類材料的提出有望擺脫目前商業陰極中對稀缺或高成本過渡金屬(如Co和Ni)的依賴。實驗進一步證實了2H-V1.75Cr0.25S4?的電壓和能量密度收益。這種策略不限于特定的無鋰陰極,并提供了一個同時實現高電壓和相位穩定的解決方案。
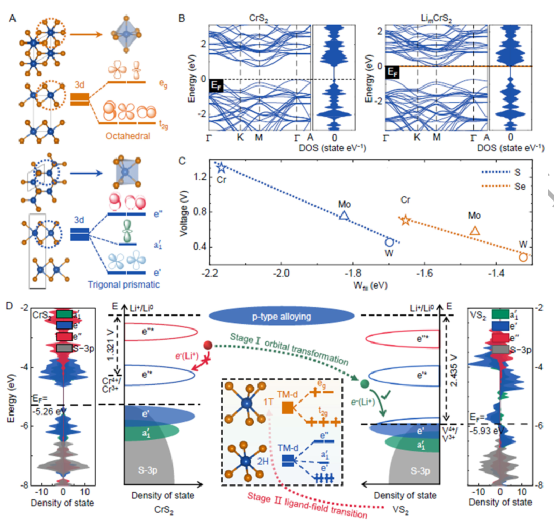
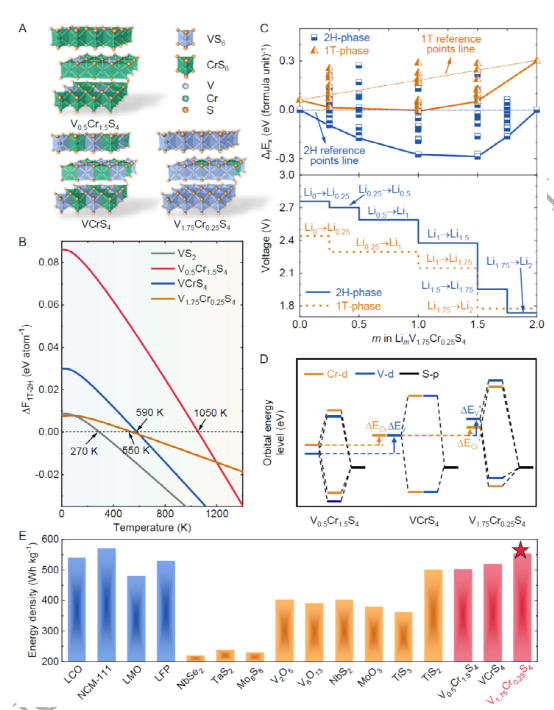
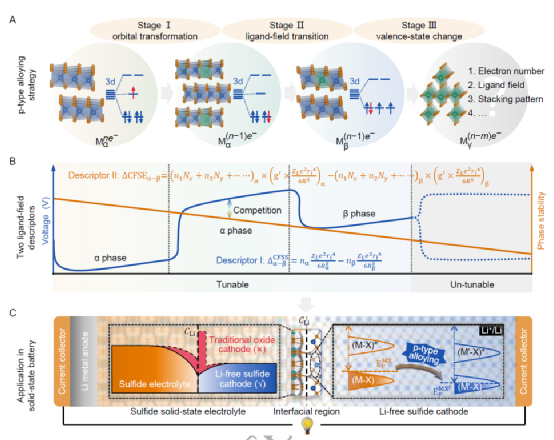
探討了陰極材料中被忽略的關鍵問題:電壓調諧與相位穩定性的競爭。提出了一種p型合金化策略,涉及三個相互聯系的階段:分子軌道轉化、配體場過渡和過渡金屬價態變化。每個制度都由兩個改進的配體場描述符,這允許調整電壓與相位穩定性的平衡并達到理想電壓。這種p型合金化策略與特定的電極材料無關。
成功地設計了一個插層型2HV1.75Cr0.25S4陰極,其初始Li+插層電壓達到2.767 V,理論能量密度達到554.3 Wh kg-1,打破了記錄。此外,2H-V1.75Cr0.25S4陰極和Li3PS4固態電解質界面提供了一個平滑的Li+遷移路徑,從而表現出比傳統氧化物電極更低的界面電阻。隨后的實驗結果證實了其優越的電壓和能量密度。
這項工作提出了通過電子帶結構工程為全固態鋰金屬電池設計硫化物陰極的定制策略,這有助于克服目前商業陰極高度依賴Co/Ni的高成本、稀缺性和集中/不穩定的供應鏈的問題。
用于穩定的全固態鋰離子電池的梯度氧-硫磷酸鹽包覆的富鎳層狀氧化物陰極

【背景】
高能富鎳層狀氧化物正極材料,如LiNi0.8Mn0.1Co0.1O2?(NMC811)在全固態鋰基電池中與硫化物固態電解質結合時,會出現有害的副反應和界面結構不穩定。
硫化物SSEs由于其在25℃時高達10?2?Scm?1?、高陽離子傳輸數(0.9)和良好的機械變形性而具有良好的前景。作為有吸引力的陰極材料,層狀氧化物陰極,特別是富含Ni的NMC陰極(例如LiNi0.8Mn0.1Co0.1O2?, NMC811),在高容量和高能量密度方面脫穎而出。然而,硫化物基ASSLBs與富鎳氧化物陰極的整合仍然遇到了嚴峻的挑戰:
1)硫化物SSEs在高電壓下的分解,因為它們的熱力學電化學穩定性窗口有限;
2)硫化物SSEs和NMC811接觸后發生界面副反應,形成離子絕緣分解產物。
3)在硫化物SSE和氧化物陰極之間形成空間電荷層(SCL),由于它們的化學電位不匹配,界面附近的Li+?離子被重新分配,導致在硫化物SSE一側形成高抗力的Li耗竭層;
4)容量和電壓衰減問題,因為結構退化發生在富鎳氧化物陰極顆粒的表面和晶界。所有這些問題應同時解決,以實現穩定和可靠的ASSLBs。
【工作介紹】
為了解決以上問題,本工作提出用鋰氧硫酸鹽(Li3P1+xO4S4x?)對NMC811顆粒進行梯度涂覆。通過Li3PO4?的原子層沉積和隨后原位形成的梯度Li3P1+xO4S4x?涂層,可以為NMC811顆粒獲得精確和保形的覆蓋。NMC811定制的表面結構和化學成分阻礙了與晶界中分層到尖晶石轉變有關的結構退化,并在循環過程中有效地穩定了陰極|固體電解質界面。
事實上,當與金屬銦負極和Li10GeP2S12?固體電解質結合測試時,基于NCM811的梯度氧硫磷涂層的正極在0.178 mA/cm2?和25℃下經過近250個循環后,能夠提供128 mAh/g的比放電容量。
【具體內容】
當硫化物SSE與具有低Li+?化學電位(μLi?)與S2-?/S的氧化物陰極材料接觸時,即使在開路電壓條件下,硫化物SSE也會被氧化,這將進一步促進SSE和陰極活性材料的結構退化。如圖1a所示,氧化物陰極材料和硫化物SSE之間形成界面,硫化物電解質側的Li+耗盡,副反應產物以及自分解產物,導致巨大的界面電阻。當構建人工氧化物界面層時,陰極|SSE界面可被視為兩個界面的組合,即陰極|界面層和界面層|SSE界面(圖1b)。
盡管與圖1a所示相比,人工界面層可以緩解Li+?的重新分布,但Li+?的貧化層仍然存在。此外,由于循環期間陰極的脫鋰/鋰化與Li+擴散相耦合,因此其高度依賴于Li+濃度和局部電勢等參數。在這方面,人工氧化物夾層不能有效緩解Li+濃度和電化學電勢的不均勻分布。
本工作基于結構和化學的高度相似性可以減少界面阻力的想法,設計了一個漸變的鋰氧-硫代磷酸酯(Li3P1+xO4S4x?)界面(圖1c),以同時保證Li+?的均勻擴散,并避免SCL的形成。同時,梯度的Li3P1+xO4S4x?界面可以保證在與硫化物SSE接觸的區域附近有較高的μLi?,避免了硫化物SSE的氧化和分解。受益于梯度的Li+?濃度、梯度的電化學電位和最小的界面電阻,還可以確保Li+?在陰極和SSE之間快速穩定的遷移。
圖1、NMC811陰極和硫化物SSE之間不同類型的相位示意圖。
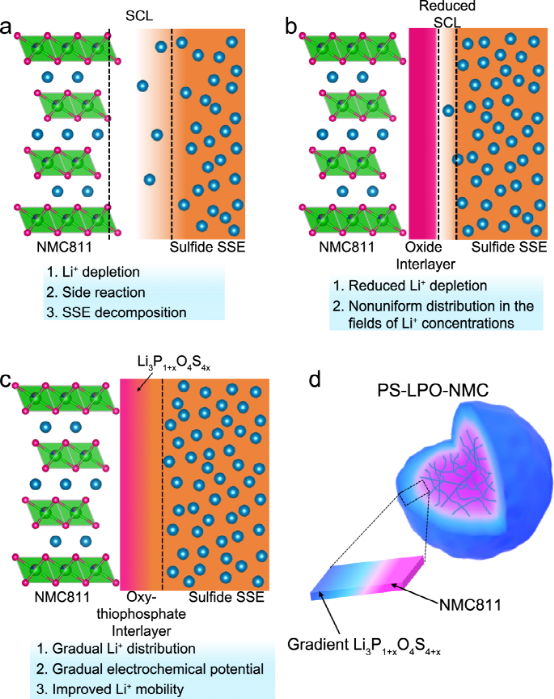
a?當無涂層的NMC811與硫化物SSE直接接觸時,形成厚的SCL;b?在NMC811上有氧化物涂層時,SCL減少;c?梯度氧化鋰-氧-硫代磷酸酯界面定制了一個從有利于氧化物到有利于硫化物的平滑過渡。d?NMC811初級粒子(粉紅色)的示意圖,帶有離子導電和梯度Li3P1+xO4S4x?涂層(藍顏色)。SCL代表空間電荷層,PS-LPO-NMC代表梯度Li3P1+xO4S4x?-涂層的NMC811。
通過對Li3P1+xO4S4x?,在NMC811原生顆粒的表面和晶界上制造一個梯度的Li3PO4?,隨后使用P4S16?輔助的固液過程進行硫化。Li3P1+xO4S4x?涂層顯示了一個漸進的濃度梯度,硫的含量向涂層的外表面增加。由于這種梯度人工涂層的離子導電但電子絕緣的性質,硫化物SSE和NMC811之間的副反應受到了阻礙。
此外,與使用氧化物涂層的方法相比,由于外部Li3P1+xO4S4x?和硫化物SSE之間的化學性質相似,因此Li3PS4- 類似的化學性質和硫化物SSE之間的電化學和化學穩定性得到改善。
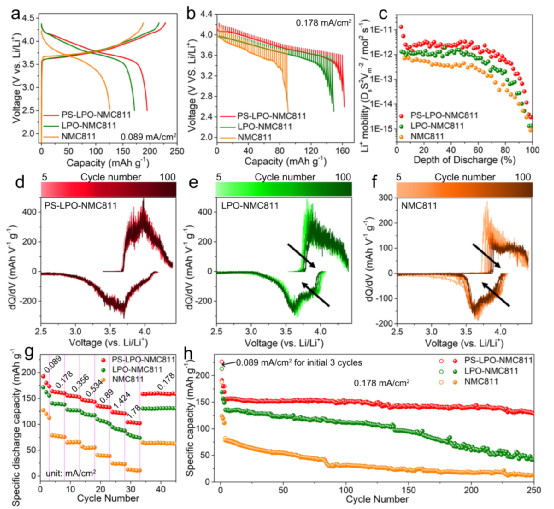
圖5、全固態鋰離子電池的電化學性能,其陽極為In,固體電解質為Li10GeP2S12,陰極為NCM811,溫度為25℃。
受益于原生NMC811顆粒表面和晶界上的梯度Li3P1+xO4S4x?涂層,Li+?離子可以順利地遷移過NMC811 |Li3P1+xO4S4x?| 硫化物SSE界面。這最終可以抑制NMC811從有利的層狀LiNi0.8Co0.1Mn0.1O2?相向不利的巖鹽Ni0.8Co0.1Mn0.1O2?相的結構轉變。因此,使用梯度Li3P1+xO4S4x?涂層的NMC811的ASSLBs在25±5℃時,在0.089mA cm?2?,實現了約160mAhg?1?的高可逆容量,與商用Li10GeP2S12?SSE集成后,250次循環后保持良好。
【結論】
提出并成功合成了一種薄而漸變的Li3P1+xO4S4x?涂層,以解決硫化物基ASSLB的高容量富鎳NMC811正極材料的循環穩定性差的問題。
通過ALD形成的Li3P1+xO4S4x?涂層,在原生NMC811顆粒的表面和晶界上實現了全覆蓋,隨后進行了原位硫化。Li3P1+xO4S4x?界面的厚度為10-20納米,在主要的無定形相中嵌入了一些結晶團。
HRTEM、同步輻射HEXPS和TOF-SIMS測量的深入分析證實了涉及富含S的Li-P-O-S的梯度成分向外表面發展,富含O的Li-P-O-S成分向陰極內部界面發展。通過梯度的Li3P1+xO4S4x?覆蓋來定制表面和晶界的結構和化學成分,穩定而快速的Li+?傳輸,可以大大減少晶界的結構退化和層狀到刺狀的轉變。
因此,陰極的容量保持和電壓穩定性得到了明顯的提高。梯度界面使In|LGPS |PS-LPO-NMC811 ASSLBs在250個循環中具有高度穩定的循環性能,放電容量保持率為80%(從第4次到第250次,在25℃下施加0.178 mA/cm2的電流)。
高壓陰極聚環氧乙烷基固態電池穩定性的評價與改進
【背景】
使用高壓陰極活性材料(CAMs)的固態電池(SSBs),如LiNi1-x-yCoxMnyO2(NCM)和聚環氧乙烷(PEO),會出現與 "波動電壓 "有關的電池故障。此外,關于它們與高壓CAMs的長期循環性能的報告并不一致。
【工作介紹】
本工作驗證了鋰枝晶通過固體聚合物電解質(SPE)的滲透確實導致這種 "波動電壓電池故障"。這個問題可以通過使用更高分子量的PEO對SPE進行簡單的修改來克服,與低分子量的PEO相比,其循環穩定性有所提高。此外,X射線光電子能譜分析證實了NCM循環后形成的氧化降解產物,由于傅里葉變換紅外光譜法的表面靈敏度有限,因此不適合作為一種分析技術。總的來說,本結果有助于嚴格評估和改善基于PEO的SSB的穩定性。
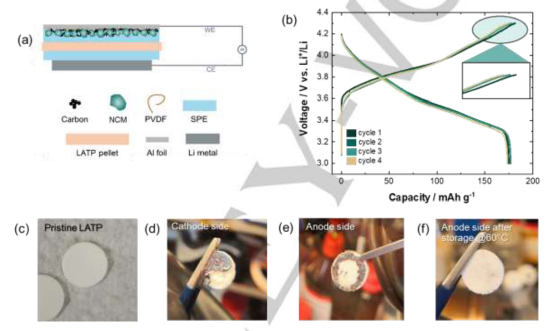
圖2.實驗結果驗證了潛在的樹枝狀穿透是導致 "電壓噪聲 "故障的原因。
通過對基于PEO的SSB與高壓陰極相結合的兼容性進行了嚴格的評估和改進。發現:鋰枝晶的形成與高壓電池的故障有關,在電池充電過程中表現為 "電壓波動",這一假設通過檢測鋰枝晶與LATP的反應產物得到證實。
為了克服這種故障,使用了分子量較高的PEO的SPE,與低分子量的PEO相比,它改善了SPE的機械剛性,使NCM具有合理的循環性能。此外,研究表明,當FTIR被用作分析技術時,經常被忽視的影響,如電池泄漏和循環過程中的溫度升高,很容易導致關于PEO氧化降解的錯誤結論。
使用像XPS這樣更敏感的表面技術,明確地證明了在SPE/NCM界面形成的氧化降解產物。對這種界面降解的詳細調查及其對電池性能的影響,取決于特定的陰極活性材料和電池電壓,將在進一步的研究中得到闡明。
可去除氫氟酸添加劑優化4.5V鋰金屬電池電極電解液與Li+導電膜的界面
【背景】
高壓鋰金屬電池(LMBs)能夠實現不斷增加的能量密度。然而,它們的循環壽命受到不穩定的電解質/電極界面和高電壓下容量不穩定的嚴重影響。
【工作介紹】
本工作提出了一種可去除氫氟酸(HF)的添加劑來優化電極電解質界面以解決上述問題。N, N-二甲基-4-(4,4,5,5-四甲基-1,3,2-二氧雜環戊烷-2-基)苯胺(DMPATMB)被用作電解質添加劑,誘導PF6??分解,形成致密和堅固的富含LiF的固體電解質界面(SEI),以抑制Li+枝晶生長。
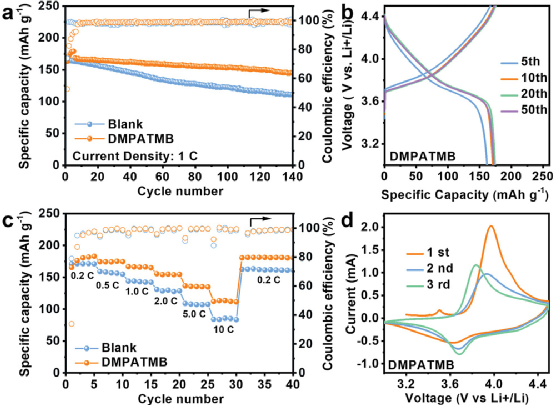
此外,DMPATMB可以幫助形成高度導電的Li3?N和LiBO2?,這可以促進Li+?在SEI和陰極電解質間相(CEI)的傳輸。此外,DMPATMB可以清除電解液中的微量HF,保護SEI和CEI免受腐蝕。正如預期的那樣,使用這種電解質的4.5VLi||LiNi0.6Co0.2Mn0.2O2?電池在200 mA g?1?的情況下循環140次后,可提供145 mAh g?1?。這項工作為LMBs的高壓電解質添加劑提供了新的見解。
【作用機制】
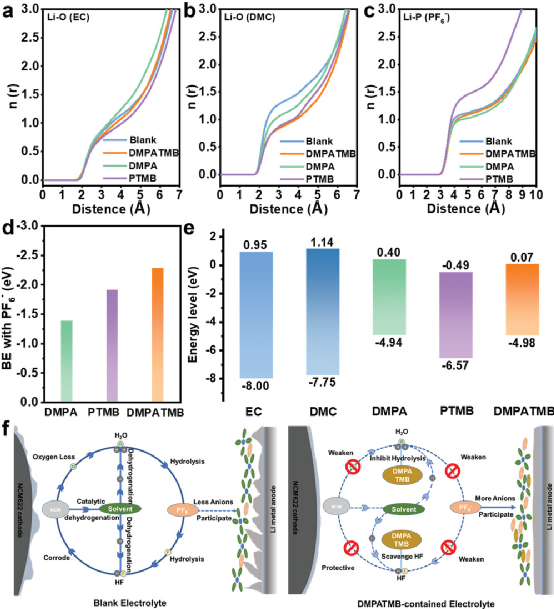
與空白電解液中的惡性循環不同,在含有DMPATMB的電解液中,電極可以得到有效保護,如圖7f所示。根據傳統的EDL理論,陰極表面的陽離子在充電時被排斥,留下大量的自由溶劑。而DMAPTMB改變了Li+?的配位,減少了Li+?- 溶劑的配位數,因此在Li陽極表面分解的溶劑較少,從而減少了有機物的含量。
此外,DMPATMB使更多的PF6??轉移到Li表面進行分解,因為它們的結合能較大。因此,在鋰陽極上形成了堅固的富含無機物、高Li+?導電的SEI,它可以有效地抑制鋰枝晶的生長,提高循環性能。在空白電解質中,溶劑在NCM622表面發生脫氫反應,產生質子,形成HF。HF可以腐蝕NCM622表面的CEI層,使陰極失去氧氣,并催化溶劑的脫氫反應,產生水。
產生的水與陰離子反應,形成更多的HF,進一步腐蝕NCM622,形成一個惡性循環。在含有DMPATMB的電解液中,DMPATMB可以清除HF,保護NCM622,并阻止隨后的副反應,減少水和HF的生成。因此,DMPATMB打破了惡性循環,形成了薄而密的CEI層。
這種CEI可以防止NCM622被腐蝕,抑制副反應的持續發生,提高電池的循環性能。同時,高Li+?導電性的CEI加速了Li+?的傳輸,從而產生了良好的速率性能。
【結論】
綜上所述,通過可移除HF的添加劑構建了高Li+?導電分子的電極電解質界面層,以維持4.5V的Li||NCM622電池的優異性能。DMPATMB可以促進LiPF6?分解,形成富含LiF的SEI,以抑制Li枝晶的生長。此外,由于DMPATMB的HOMO和LUMO值適中,它參與了富含Li3N和LiBO2?的SEI和CEI的形成。
此外,DMPATMB可以清除HF和抑制PF6?的水解,以防止電極材料的腐蝕。DMPATMB的這些優點賦予了Li||NCM622電池在4.5V電壓下卓越的循環穩定性和速率性能。這項工作不僅提供了一種用于4.5V鋰電池的多功能添加劑,而且為設計高壓LMB的功能添加劑提供了新的思路。
有機硫鋰電池用聚苯胺骨架硒摻雜正極材料
【背景】
盡管鋰硫電池具有超高的比容量,但由于可溶性多硫化物(LiPSs)從陰極到陽極自由穿梭,導致活性物質的損失和不可逆的容量衰減。此外,差的絕緣性能導致了反應動力學的遲緩。由于不可逆的容量衰減和緩慢的反應速度,阻礙了鋰硫電池的實際應用。
【工作介紹】
本工作采用了質子酸摻雜的硫化聚苯胺(SPANI)作為正極的活性物質,它具有多個質子化的醌類亞胺(-NH+= 、-N =),在-NH+?=質子化的醌類亞胺和去質子化的醌類亞胺(和)之間做可逆轉換,在電化學反應中同時吸附和解吸LiPSs。有趣的是,這種由LiPSs和Se引發的可逆轉化作為成功的摻雜原子,增強了電子傳導性,提高了鋰離子傳輸率。
作者設計了摻硒的SPANI作為陰極活性材料,揭示了其反應機制。引入Se有幾個優點:
i)Se確實更容易取代S,因為Se是周期表中的一組VIA元素,也是S的同系物,它有許多與S相似的特性;
ii)Se的電子傳導率(1×10-3?S m-1)比S(5×10-28?S m-1)高得多,允許活性材料具有更強的電子傳導率;
iii)Se的電負性比S低,因此將Se與S結合會改變S的狀態密度。在PANI與SexS的退火過程中,一個S原子將被Se原子取代,由于它們的電負性不同,改變了相鄰S原子的軌道能級分布。同時,它將獲得存在于Li+?s和S2-?sp3之間的更窄的能隙,從而加速了低階多硫化鋰,Li2S和Li2Se的快速成核。
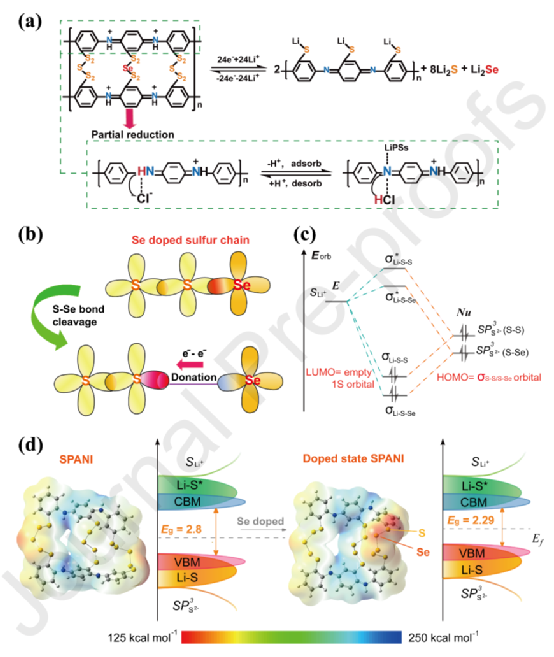
摻入Se的硫化聚苯胺與調整后的硫鏈-Sx- (x?≤ 6)有助于確保沒有長鏈多硫化物,而帶有醌類亞胺的骨架可以通過去質子化/質子化亞胺(-NH+=和-N=)之間的可逆轉換賦予對短鏈多硫化物的強烈吸附,這為抑制 "穿梭效應 "提供了雙重保險。
此外,在硫化多硫化物中摻入Se原子,以加速氧化還原的轉換,通過前線軌道理論為導向,研究了催化機制。在軌道能級圖中,Se0.07SPANI的單鋰化能隙為2.29 eV,比SPANI的2.8 eV低。由于摻入Se并與S形成共價鍵將調控S的電子密度差,導致電位降低,費米級更接近S中心的Sp3?帶。該策略降低了固-固反應的勢壘能,加速了電極反應的動力學。
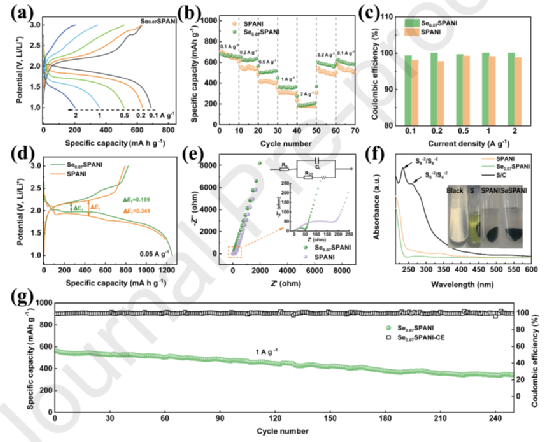
設計的Se-摻雜的硫化聚苯胺作為鋰-有機硫電池的正極活性材料,表現為一種新式的 "固-固 "反應機制,沒有可溶性LiPSs的穿梭,這一點通過DFT計算和紫外可見光譜學得到了揭示。這是因為Se0.07SPANI的短硫鏈(?Sx??, x ≤ 6)可以保證沒有可溶性的長鏈LiPSs,同時去質子化的醌類亞胺功能可以與可溶性LiPSs強烈吸收。
同時,所采用的Se-摻雜改變了雜化軌道的帶能分布,形成了穩定的?Li-Se鍵和靠近中心S sp3帶的費米水平。該策略降低了固-固反應的勢壘能,加速了電極反應的動力學。該工作不僅選擇了新的優勢SCP作為活性材料,解決了 "穿梭效應 "問題,而且采用了摻入Se的策略,提高了反應速率,取得了優異的電化學性能,為鋰-有機硫電池的化學導向結構設計和實際應用提供了寶貴的指導思想和有前景的解決方案。
電結晶調節使堆積的六邊形板塊向高可逆鋅陽極生長
【背景】
實現持久的平面,無枝晶的鋅(Zn)金屬構型是解決由內部短路引起的電池過早失效的關鍵,這在很大程度上是由電結晶過程中的晶體生長決定的。
【工作介紹】
本工作通過調節內亥姆霍茲平面(HIP)的分子結構,通過削弱界面上的溶劑化離子吸附,有效地將沉積轉化為活化控制。緩和的電化學反應動力學低于原子自擴散速率,引導符合要求的層狀鋅生長和主導的鋅(0001)面,實現了晶體學的優化。通過電解質工程的原位調解,邊緣部位的定向電鍍和剝離行為以及定制的溶膠結構極大地提高了金屬鋅的利用效率和總電荷量,即使在極端條件下,包括高面積容量(3 mAhcm-2)和寬溫度范圍(-40~60℃)。
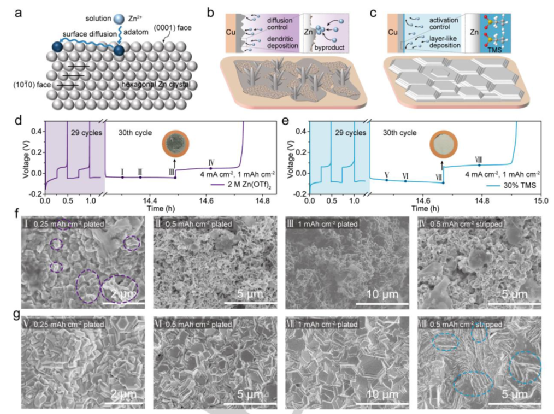
通過采用環狀和線狀砜(即TMS和DMSO)作為特定對象,證明了特異性吸附的分子是如何調控電結晶的,研究證明了控制界面上的溶質離子吸附以延緩電化學反應動力學低于原子自擴散速率,導致首選取向的重要性。IHP中TMS垂直偶極陣列的形成,外環結構的低電子密度,以及親電位點的巨大立體阻礙,有效地減弱了與溶劑化離子的相互作用,作為一個新的限速步驟,將沉積轉化為活化控制。
充分的原子自擴散促進了橫向的、逐層的生長,使(0001)面暴露出來,產生了由堆疊的六邊形板塊形成的平坦而密集的鍍層。結合KPFM,表明剝離點與生長點相吻合,這進一步確保了循環過程中形態和面的穩定性和可重復性。此外,由于溶劑化結構和氫鍵網絡的重建,TMS的引入可以抑制氣體的產生。
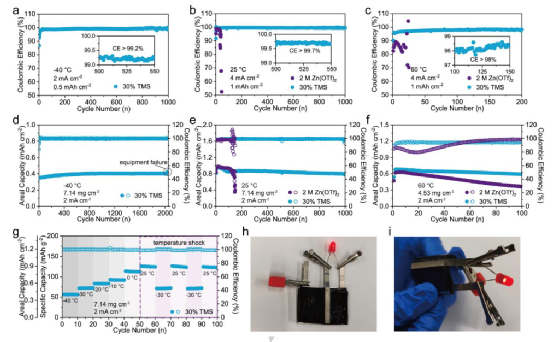
所設計的電解質有助于明顯提高ZnCE(1000次循環中大于99.7%,提高33倍以上)和Zn/PANI全電池的性能(1000次循環的穩定循環,保留率為85%)。更值得注意的是,在-40°C和60°C的極端溫度下,性能的提高變得非常突出。
研究結果深入了解了添加劑通過特定的吸附作用對電沉積結構的影響,以及在電解質設計方面的新見解,以在原位構建結晶學面、無樹枝狀物的鋅陽極。
審核編輯:劉清





















 工商網監
工商網監
評論