 電子發燒友App
電子發燒友App


? 一、背景介紹
開發先進的電解液是開發下一代鋰離子電池(LIBs)不可或缺的重要組成部分。然而,Li+與各種溶劑之間的強溶劑化相互作用,往往導致Li+去溶劑緩慢和溶劑共嵌到石墨電極中,從而極大地限制了電解液設計。相比之下,非配位共溶劑(非溶劑)對Li+保持惰性,但由于較小的非溶劑-溶劑相互作用,將體相溶劑化網絡分離為單個溶劑簇,為調整Li+-溶劑的強度和拓撲結構鋪平了另一條途徑。
此外,理想的非溶劑可以改善物理性能(如粘度、共晶溶劑和潤濕性),并為電解液設計激發一個新的維度。因此,探索這些分子相互作用(如Li+-溶劑吸引和非溶劑-溶劑相互作用)和描述它們的臨界條件對于開發多功能電解液和調整其電化學行為是勢在必行的。
之前的研究中,華中科技大學謝佳教授和曾子琪博士等人(-90~90°C!非溶劑化相互作用實現寬溫域PC基電解液)在Adv.Energy Mater.上以“RejuvenatingPropylene Carbonate-based Electrolyte?Through NonsolvatingInteractions for Wide-Temperature Li-ionsBatteries”為題提出了通過在PC和鋰鹽構成的PC基電解液中加入氟苯(FB),基于非溶劑化的相互作用來調節Li+-PC的強度和拓撲結構,使得PC溶劑寬的溫度范圍(-90~90°C)內實現石墨負極鋰離子全電池的循環。
作者認為,在2.3M的較低鋰鹽濃度下,FB能夠削弱PC和Li+之間的親和力,而溶劑化結構沒有顯著變化,電化學穩定性增強,且高低溫電化學性能明顯提高。
針對DME、DMSO、TMP、PC和DEC等一類具有強溶劑化效應的溶劑,本文提出的非溶劑化相互作用來調節Li+的溶劑化結構是否也適用于此呢?
二、正文部分
01 成果簡介
在此,華中科技大學謝佳教授和曾子琪博士等人提出了偶極-偶極相互作用的機制,通過調節Li+、溶劑和非配位分子(非溶劑)之間的相互作用來促進Li+去溶劑和抑制溶劑化Li+共嵌。
具體而言,在中等鋰鹽濃度下,非溶劑抵消靜電吸引,以抑制Li+和溶劑之間的親和力,而不改變初級溶劑化結構。因此,Li+溶劑化強度減弱,使去溶劑化變得容易,從而與石墨負極在各種溶劑中具有優異的電化學相容性,包括DME(乙二醇二甲醚)、DMSO(二甲基亞砜)、TMP(磷酸三甲酯)、PC(碳酸丙烯酯)和DEC(碳酸二乙酯)。
因此,本文提出的偶極子-偶極子相互作用的策略可以將電解液設計的視野擴展到先進的LIBs中。該研究以題目為“Dipole-dipoleInteractions for Inhibiting Solvent Co-intercalation into GraphiteAnode to Extend the Horizon of ElectrolyteDesign”的論文發表在材料領域國際頂級期刊《EnergyEnviron. Sci.》。
02 研究亮點
1.本文提出了一個分子相互作用模型來調節溶劑化Li+的界面行為,非溶劑遠離了Li+的溶劑化殼層,同時對極性溶劑產生了相當大的偶極-偶極相互作用,削弱了Li+與溶劑之間的庫侖吸引力;
2.松散的Li+-溶劑團簇在熱力學上有利于有效的去溶劑化,從而降低了去溶劑化勢壘,改善了電化學穩定性
3.在中等鋰鹽濃度(臨界邊界條件)下,利用各種非溶劑(FB、ClB、BrB和CB),基于其他正極不穩定溶劑(如DME、DMSO、TMP、PC和DEC)驗證了該模型。
03 圖文導讀
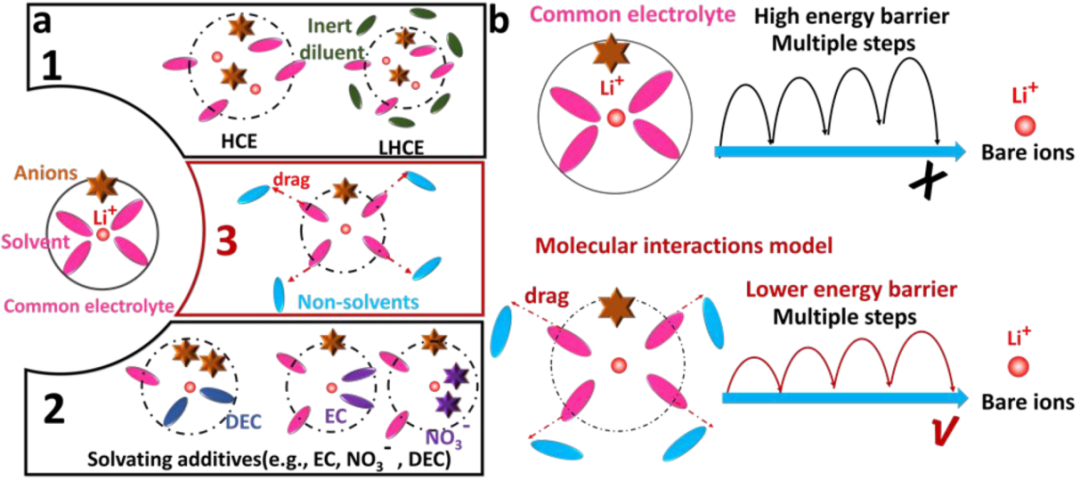
【方案1】已建立的溶劑化結構及去脫溶劑過程的示意圖。(a-1)高濃度電解液和局部高濃度電解液的溶劑化化學性質;(a-2)具有不同溶劑化組分(如EC、LiNO3和DEC)的電解液的溶劑化化學性質;(a-3)加入非溶劑后的電解液的溶劑化化學性質和分子相互作用模型;(b)普通電解液和非溶劑改性電解液的其去溶劑化機理。
1.電解液成分的合理設計
在低濃度摩爾比(LiFSI/DME<1:4)時,石墨//Li電池表現出可逆共嵌;當LiFSI/DME=1:3(LiFSI-3DME)時,可以獲得更高的220mAh g-1可逆容量(圖1a)。相比之下,LiFSI-2DME粘度的急劇增加,表現出高極化和低容量。值得注意的是,LiFSI/DMEMRs的增加在一定程度上抑制了Li+-DME共嵌,但仍存在高粘度、低電導率和殘留共嵌(0.8V平臺)。
在LiFSI-3DME體系中引入氟苯(FB),共嵌被完全抑制,石墨的理論容量高達372mAhg-1,初始庫侖效率高達86%(圖1b)。此外,這種電解液(LiFSI-3DME-7FB)在石墨//Li電池中顯示出超過200次循環的穩定循環,容量保持率高達94%(圖1c)。
OperandoXRD顯示LiFSI-3DME電解液中顯示出明顯的(002)峰分裂(圖1d)。相比之下,在FB支持的DME電解液中,石墨在0.3V以上保持(002)峰,表明在Li+嵌入之前,抑制了Li+-DME共嵌和穩定了層狀結構。CV結果進一步驗證了引入FB后DME基電解液相容性的改善(圖1e)。
值得注意的是,不同于常見的HCEs和LHCEs,應該開發一種新的模型來解釋僅包含鋰鹽、溶劑和FB的電解液設計中存在的抑制共嵌行為。
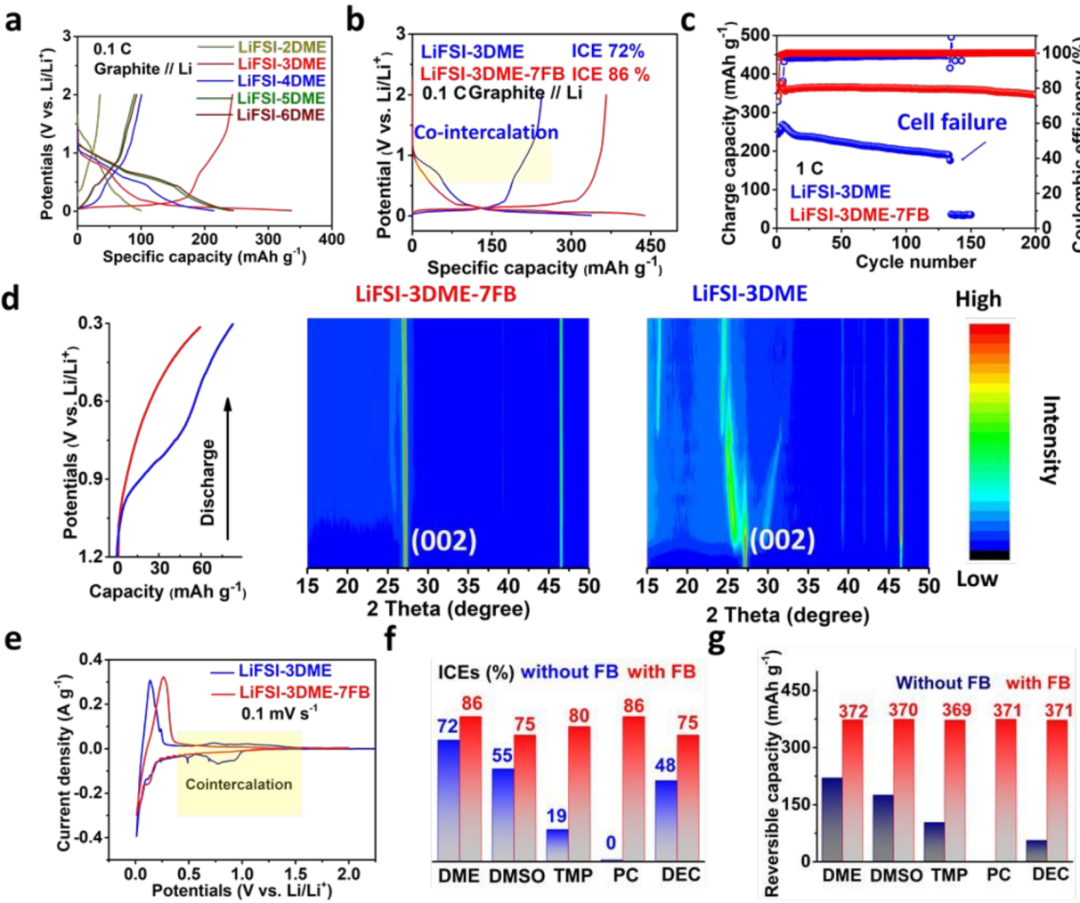
【圖1】溶劑共嵌表征。(a,b)不同電解液中石墨/Li電池的充放電曲線;(c)石墨/Li電池的循環穩定性;(d)原位XRD表征;(e)LiFSI-3DME和LiFSI-3DME-7FB電解液中石墨/Li電池的CV曲線;(e,f)加入FB后ICEs和可逆容量的比較。
2.模型通用性演示和溶劑到非溶劑的選擇
為了揭示電解液的設計和建立分子相互作用模型,本文系統地研究了DMSO、TMP、PC和DEC在石墨//Li電池中的陰極穩定性。值得注意的是,石墨在低濃度下存在嚴重的共嵌,但隨著鋰鹽/溶劑MRs的增加,相容性逐漸增強。因此,根據放電曲線,LiFSI-3DMSO(4.7M)、LiFSI-3TMP(3.1M)、LiFSI-5PC(2.3M)和LiFSI-6MEC(1.4M)為臨界摩爾比,高MRs下可逆循環,低MRs下電解液失效。
當FB加入后,電化學可逆性行為,ICEs分別增強為75%、80%、86%和75%。此外,在石墨相容溶劑(如EMC、DMC和EC)中也研究了FB的效應,與原始電解液相比,其倍率性能大大提高。
同時,考慮到FB的脫氟可能促進初始循環過程中石墨表面含LiF的SEI的形成,作者使用其他無F原子的非溶劑(如ClB、BrB和CH)取代FB。含ClB和BrB的電解液的電化學相容性和可逆性顯著增強,其高ICEs分別為85.9%和86.6%(圖2a,b)。更重要的是,僅含C和H原子的環己烷(CH)在LiFSI-5PC-7CH中也顯示了石墨的可逆循環(圖2c)。
以上結果表明,非溶劑并不局限于FB,還可以擴展到其他非溶劑,如ClB、BrB和CH(圖2d),證明了該設計原理的通用性。此外,含FB的電解液保證了在SEI較差的情況下石墨的正常運行,進一步說明了電解液結構和偶極-偶極相互作用的主導作用。
此外,其他報道也揭示了SEI對抑制溶劑共插層的有限作用,突出了體相電解液設計的意義。這些結果表明:(i)非溶劑的影響不是由其成膜能力引起的;(ii)體相電解液或Li+-溶劑配位可能比SEI對抑制Li+-溶劑共嵌的作用更大。
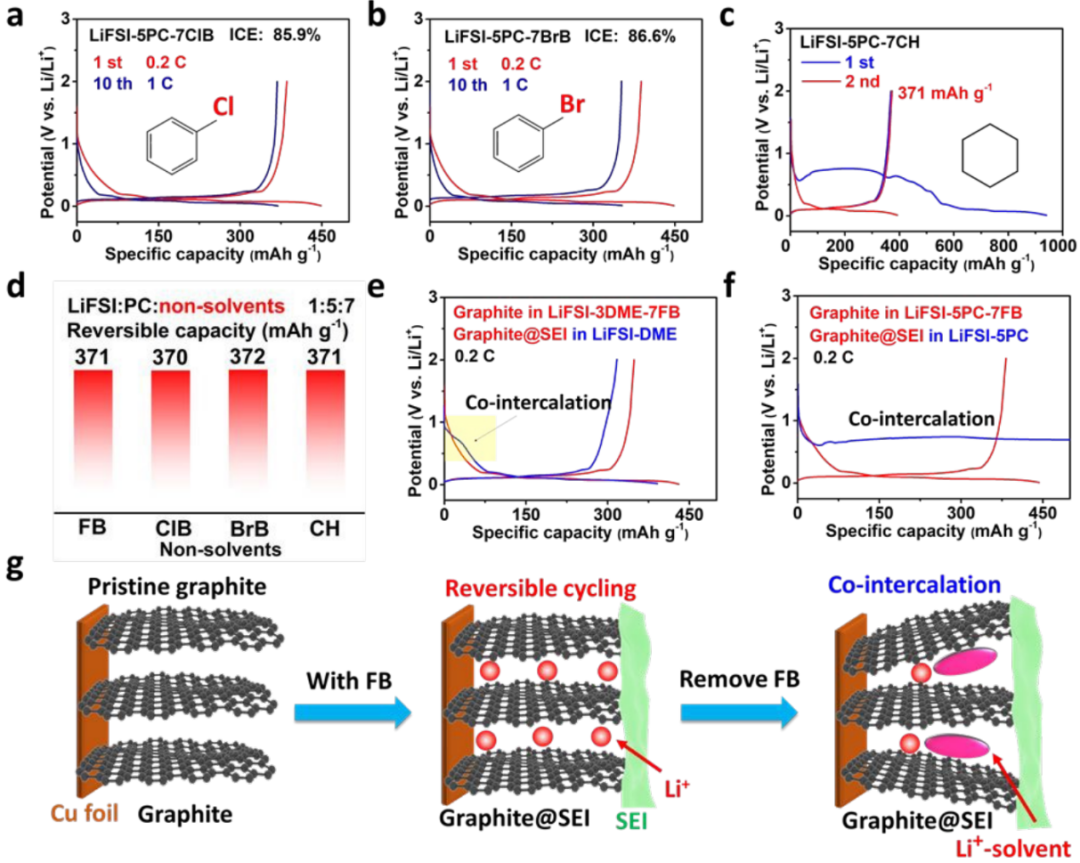
【圖2】SEI非溶劑的選擇及抑制共嵌的效果驗證。(a-c)Li-FiSI-5PC-7ClB、LiFSI-5PC-7BrB和LiFSI-5PC-7CH中石墨/Li電池的充放電曲線;(d)加入不同非溶劑后的可逆容量;(e,f)SEI涂層石墨在無FB的電解液中重新組裝;(g)含FB和無FB的電解液中石墨電極的原理圖。
3. 分子相互作用和溶劑化結構
為了確定與石墨的相容性增強的關鍵因素,利用核磁共振(NMR)和拉曼分析研究了鋰鹽、溶劑和非溶劑的相互作用。與FB混合后,DME中1H核的信號從3.97ppm轉移到3.41ppm,與FB中1H核從6.90ppm下降到7.12ppm完全一致,表明FB與DME之間存在分子相互作用(圖3a)。
在DME中LiFSI解離后,DME的1H信號從3.97ppm移動到3.78ppm,表明DME和Li+之間具有很強的庫侖吸引力(圖3b)。將FB引入到LiFSI-3DME電解液中,由于Li+-DME與FB-DME之間的累積相互作用,導致DME中1H核的進一步上升(圖3c)。
結果表明,分子尺度競爭模型顯示Li+-DME團簇具有較強的庫侖吸引力,而FB-DME具有相對較弱的相互作用(偶極-偶極相互作用)。此外,在與PC、DMSO和TMP的電解質中也觀察到類似的分子相互作用,表明在體相電解液中普遍存在分子規模的競爭。
同時,通過拉曼數據揭示了溶劑化結構,CIPs和AGGs的增加(從46%增加到53%)不足以形成陰離子衍生的SEI,而通過重新組裝SEI涂層石墨顯示,SEI對抑制共嵌的作用有限(圖2e-g)。
FB與DME之間的偶極-偶極相互作用明顯弱于Li+與DME之間的庫侖吸引,導致Li+-DME強度降低,但保留了溶劑化結構,如MD模擬所示(圖3f)。基于以上討論,LiFSI-3DME和LiFSI-3DME-7FB的溶劑化結構,在前者中,極性溶劑(如DME和PC)與Li+之間的高親和力有助于產生較強的庫侖吸引力,導致了穩定和緊湊的溶劑化結構(圖3g)。
當引入FB時,FB和DME之間的偶極-偶極相互作用部分削弱了Li+-DME的吸引力,構建了一個相對松散的溶劑化結構,而沒有明顯排擠DME分子(圖3h)。
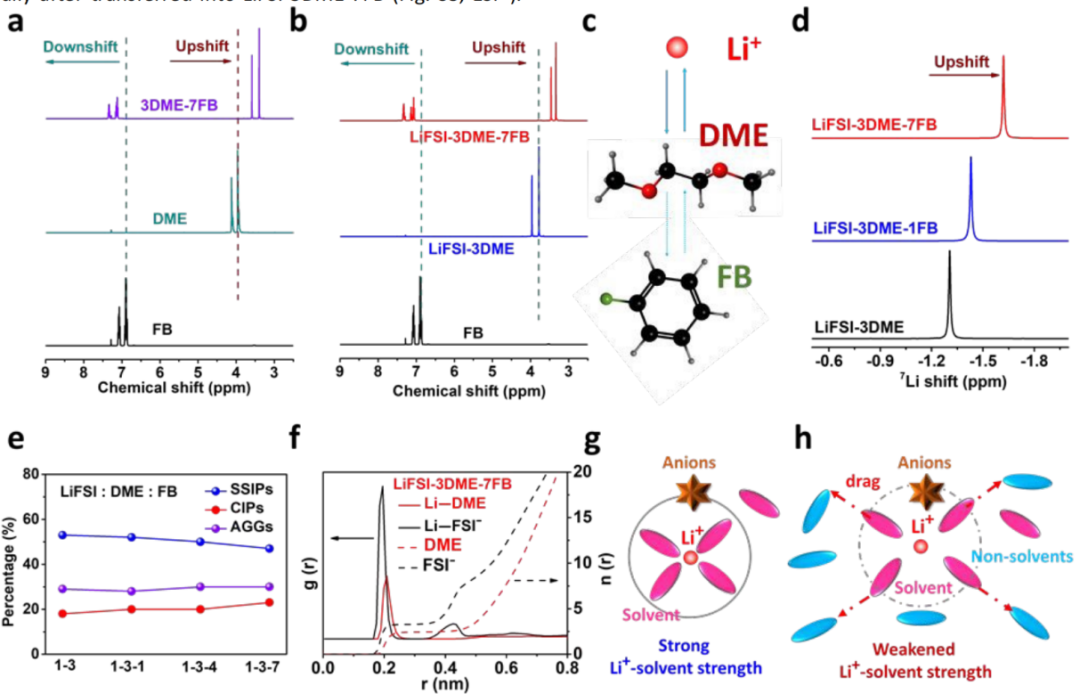
【圖3】非配位的相互作用和溶劑化結構。(a)FB、DME和混合溶液的1H信號;(b)FB、LiFSI-3DME和LiFSI-3DME-7FB信號;(c)FB、PC和Li+之間的相互作用;(d)各種電解液的7Li信號;(e,f)添加FB后的SSIPs、CIPs和AGGs的百分比,以及游離DME和Li+-DME的百分比;(g,h)LiFSI-3DME和LiFSI-3DME-7FB的溶劑化結構示意圖。
4.去溶劑化和界面行為
實驗表明,去溶劑化作用主要在電極/電解液界面上進行,并決定了電化學行為。FB降低了DME基電解液中-3.44到-1.23eV的結合能,以及PC基電解液中-2.73到-0.71eV的結合能,表明Li+-溶劑化能力的減弱(圖4a)。同時,還計算了均方位移(MSD)隨時間的變化,以了解相對松散的溶劑化殼層和相對降低的Li+-DME強度。加入FB后,Li+的擴散系數從7.87×10-9增加到
相比之下,原始電解液(如LiFSI-3DME、LiFSI-5PC)具有強結合和緊密的配合,具有緩慢的去溶劑化和共嵌發生(圖4d)。此外,FB、PC和DME在石墨(001)平面上的吸附能分別為-0.513、-0.464和-0.817eV,FB的吸收能力強于PC,但略低于DME。然而,LiFSI-3DME-7FB中較高的FB含量補償了這種吸收能隙,使得FB在石墨表面的積累(圖4e)。加入FB后,石墨表面附近DME的DME頻率明顯降低,導致DME還原的可能性較小(圖4f,g)這些結果表明,FB削弱了Li+-溶劑的吸引力,保護了溶劑在石墨表面附近的電子交換,從而增加了電化學相容性,添加FB也降低了粘度,增加了潤濕性。
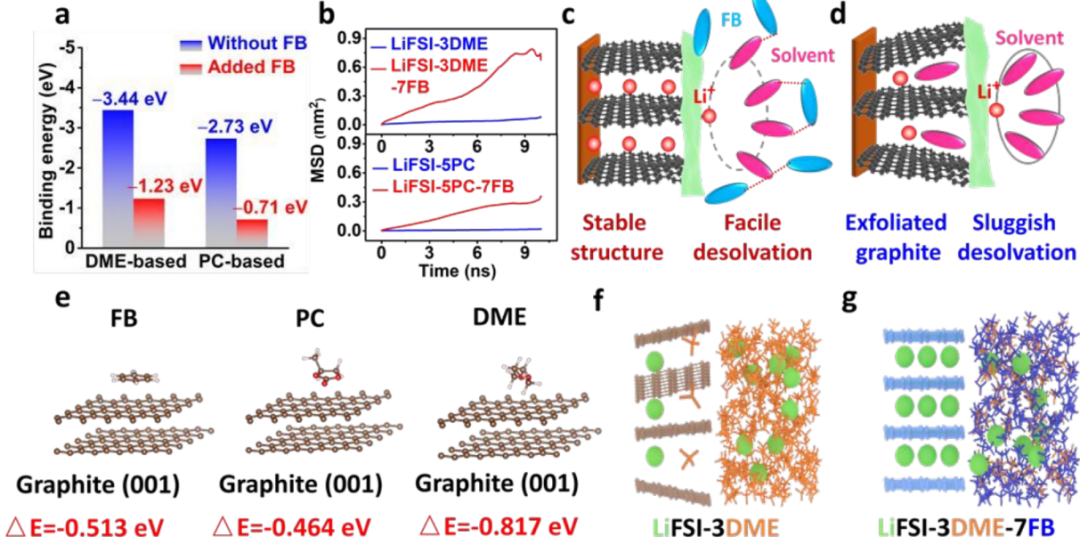
【圖4】添加FB后的去溶劑化和界面機理。(a)加入FB前后Li+-溶劑的結合能;(b)MSD在DME和PC電解液中與時間的函數;(c,d)LiFSI-3DME-7FB和LiFSI-3DME在石墨表面去溶劑的插圖;(e)FB、PC和DME在石墨(001)平面上的吸附能;(f,g)LiFSI-3DME和LiFSI-3DME-7FB中的界面狀態示意圖。
5.分子相互作用模型和關鍵因素
基于以上討論,建立了分子相互作用模型來解釋鋰鹽/溶劑/非溶劑的界面行為和臨界MRs,該模型的本質在于Li+-溶劑(受鋰鹽濃度和溶劑類型的影響)和非溶劑-溶劑相互作用(受極性和非溶劑含量的影響)(圖5a)。競爭力決定了去溶劑的能力,從而影響了電化學性能。強大的Li+-溶劑吸引力需要更強的非溶劑-溶劑相互作用來抵消和提供可行的去溶劑化,反之亦然。
結果表明,LiFSI-4DME-7FB和LiFSI-6PC-7FB不能在石墨//Li電池中發揮作用,在較低的鋰鹽/溶劑中,溶劑化殼層內主要的Li+溶劑團簇很難被偶極-偶極相互作用削弱(圖5b)。此外,臨界Li鹽/溶劑MRs依賴于Li+-溶劑的結合能(溶劑類型),由于螯合作用,DME表現出的結合強度最強,其次是PC和DEC(圖5c,d)。
具有最高結合能的Li+-DME團簇更難被分子相互作用所取代,需要更高的Li鹽/溶劑的臨界MRs(LiFSI-3DME)來降低Li+-DME的強度(圖5b)。相比之下,PC和DEC可以在相對較低的Li鹽/溶劑的臨界MRs下工作。簡而言之,鋰鹽/溶劑的臨界MRs依賴于溶劑,并影響Li+-溶劑的相互作用。
對于偶極-偶極相互作用,含量和極性是兩個關鍵因素。隨著DME和PC中FB含量的增加,ICEs含量越高(圖5e)。此外,對選定的非溶劑的電子云模擬中,CH具有對稱的環狀結構,呈現出平均分布的電子云。相比之下,由于鹵素元素電負性較高的BrB,FB、和ClB分別在F、Cl和Br原子周圍存在電子云堆積,可以合理地推測BrB-溶劑對的相互作用最強,因此提高石墨相容性所需的非溶劑含量最低(圖5g)。
結果證實,建立非溶劑的有效值為LiFSI-5PC-1BrB、LiFSI-5PC-4ClB和LiFSI-5PC-7FB,與非溶劑的極性一致(圖5f)。然而,環己烷具有最小的極性,即使在LiFSI-5PC-7CH的初始階段也提供了最低的庫侖效率,但仍能保持可逆循環(圖2c)。
因此,Li+-溶劑和溶劑-非溶劑之間的微妙平衡可以通過這些途徑來調整:1)根據溶劑的溶劑化能力調整鋰鹽/溶劑的摩爾比;2)利用高度極化的分子來增強偶極-偶極相互作用;3)在合理的范圍內增加非溶劑的含量,以加強偶極-偶極相互作用。
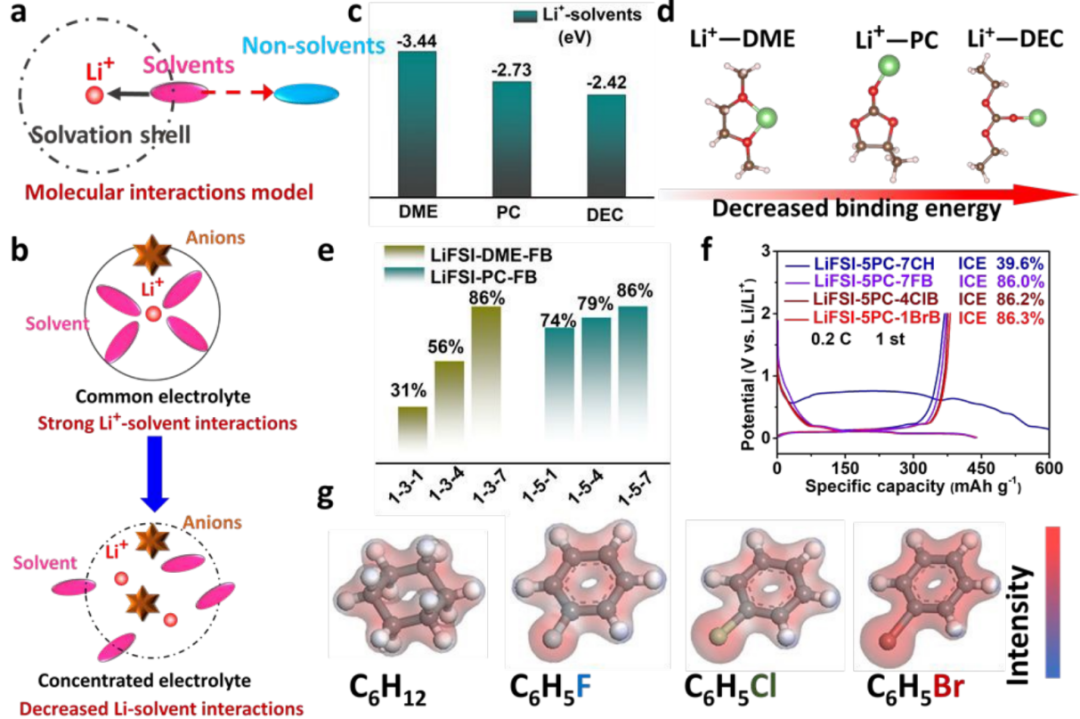
【圖5】分子相互作用的模型和機制。(a)分子相互作用模型的示意圖;(b)增加鋰鹽/溶劑MRs后,Li+-溶劑相互作用降低;(c)計算得到的Li+-溶劑結合能;(d)DME與PC和DEC的結合強度降低;(e)隨著FB含量的增加,ICEs增加;(f)獲得理想的ICEs所需的非溶劑含量;(g)不同非溶劑的模擬電子云。
6.所設計的電解液的性能指標和實用性
本文選擇LiFSI-3DME-7FB和LiFSI-5PC-7FB作為原型,以證明這種分子相互作用模型在電解液中的實用性。結果表明,對于基于DME(LiFSI-3DME-7FB)和基于PC(LiFSI-5PC-7FB)的電解液的離子電導率均有所提高(圖6a),其電解液的電化學窗口提升明顯(圖6b)。
因此,在鋰鹽、溶劑和非溶劑的合理配方下,電解液可以表現出多功能性能(如低溫適應性、高導電性、高壓耐受性)。此外,LiFePO4//石墨軟包電池在LiFSI-3DME-7FB中可提供可逆的充電放電,其ICE高達92%(圖6c)。最重要的是,PC基電解液在1.6AhLiFePO4//石墨軟包電池中被評估,獲得了91%的高ICE(圖6d),表明在FB的協助下,其對LiFePO4和石墨都具有良好的相容性。
此外,軟包電池可以在0.5C可逆循環160次循環,容量衰減小,表明PC基電解液在實際條件下的可靠性。
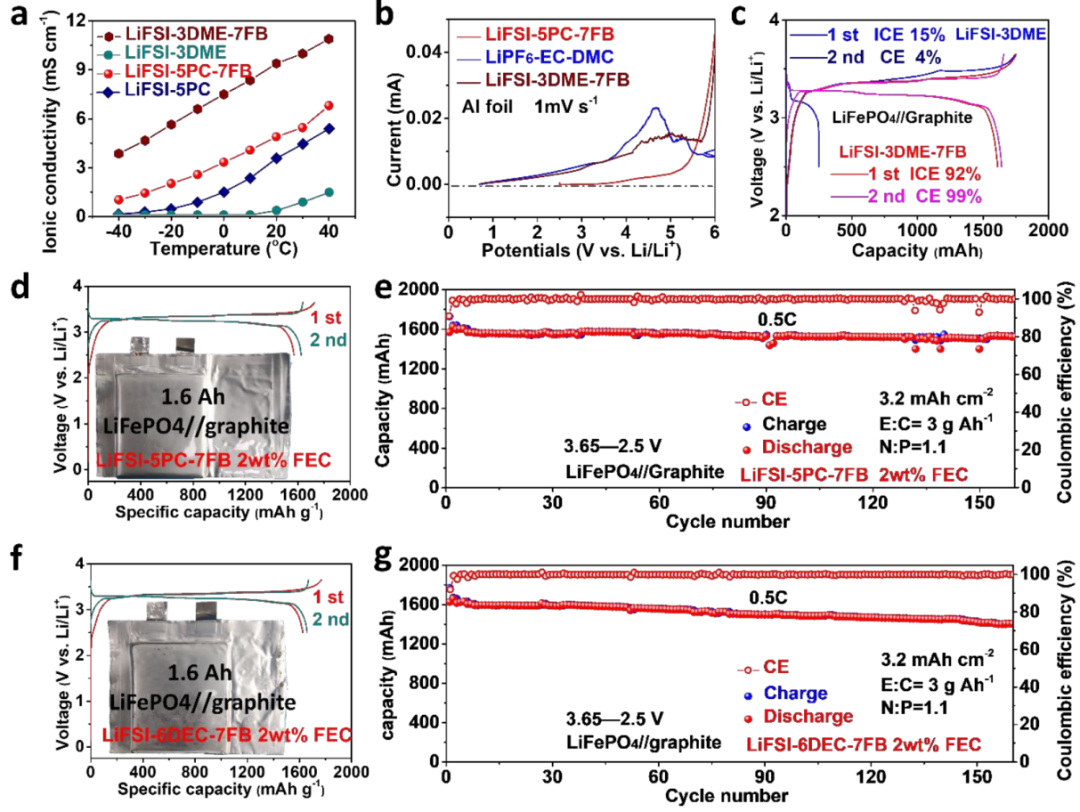
【圖6】電解液的特征和應用。(a)PC基和DME基電解液在寬溫范圍內的電導率;(b)LSV測試結果;(c)DME基電解液中LiFePO4/石墨軟包電池的充放電曲線;(d,e)LiFePO4//石墨軟包電池在LiFSI-5PC-7FB電解液中的初始充放電曲線和循環穩定性;(f)LiFePO4//石墨軟包電池在LiFSI-6DEC-7FB電解液中的初始充放電曲線和循環穩定性。
? 4、總結和展望
綜上所述,本文提出了偶極-偶極相互作用模型,用于增強各種電解液與石墨負極的相容性,抑制了Li+-溶劑共嵌,保證了石墨晶格和增強了電化學可逆性。在該模型中,將非溶劑(如CH、FB、ClB和BrB)以臨界鋰鹽/溶劑摩爾比引入溶劑(如DME、PC、DEC、TMP、DMSO)中,使Li+與溶劑的親和力減弱,表面去溶劑和石墨相容性增強,而不改變初級溶劑化結構。該模型的本質取決于Li+溶劑與非溶劑的相互作用,兩者決定了去溶劑動力學并決定了電化學行為。分子相互作用模型為提高中等鋰鹽濃度下多功能電解液與石墨負極的界面穩定性提供了一種新的策略,為優越的鋰離子體相電解液設計提供了思路。
審核編輯:劉清


















 工商網監
工商網監
評論