 電子發燒友App
電子發燒友App


?
研究背景
鈉金屬電池是一種具有高能量密度和低成本的電池,在能源存儲領域具有廣泛的應用前景。構建富含無機物且堅固的固體電解質界面(SEI)是提高鈉金屬電池(SMBs)電化學性能的關鍵方法之一。然而,常見無機物在SEI中的低導電性和分布會擾亂Na+擴散并引起不均勻的鈉沉積,導致鈉金屬電池的循環壽命和安全性能受到限制。因此,研究人員一直在尋找新的方法來解決這些問題,實現高性能和長壽命的鈉金屬電池。
成果介紹
近日,北京化工陳仕謀、鄭州大學陳衛華教授團隊介紹了一種新的方法來構建應用于鈉離子電池的固體電解質界面(SEI),通過引入自我犧牲的LiTFSI到鈉鹽基碳酸鹽電解液中,構建了一種具有高導電性無機物均勻分散的獨特SEI,這種SEI方法實現了超高速率和長壽命的鈉金屬電池制備。這種方法基于還原競爭效應,利用LiTFSI和FEC衍生的SEI使Na∥Na3V2(PO4)3電池在高達60 C的超高速率下經過10000個循環后仍保持89.15%的容量保持率。這種獨特的SEI方法為超高速SMBs提供了一種簡單、經濟和高效的界面設計途徑。
該工作以“Reductive Competition Effect-Derived Solid Electrolyte Interphase with Evenly Scattered Inorganics Enabling Ultrahigh Rate and Long-Life Span Sodium Metal Batteries”為題發表在“Journal of the American Chemical Society”上
研究亮點
(1) 介紹了一種新的方法來構建堅固且富含無機物的固體電解質界面(SEI),通過這種方法,實現了超高速率和長壽命的鈉金屬電池。
(2) Na∥Na3V2(PO4)3電池在高達60 C的超高速率下經過10000個循環后仍保持89.15%的容量保持率。
(3) 這種獨特的SEI方法在開發高性能鈉金屬電池方面具有潛在的應用前景。
圖文導讀
一 電解質的物理化學性質
首先對電解質的物理化學性質進行研究,以驗證LiTFSI在電解質中的有效性。為了揭示鹽濃度和溶劑對電解質離子導電性的影響,選擇了不同濃度的主要鹽NaClO4在FEC和PC中。
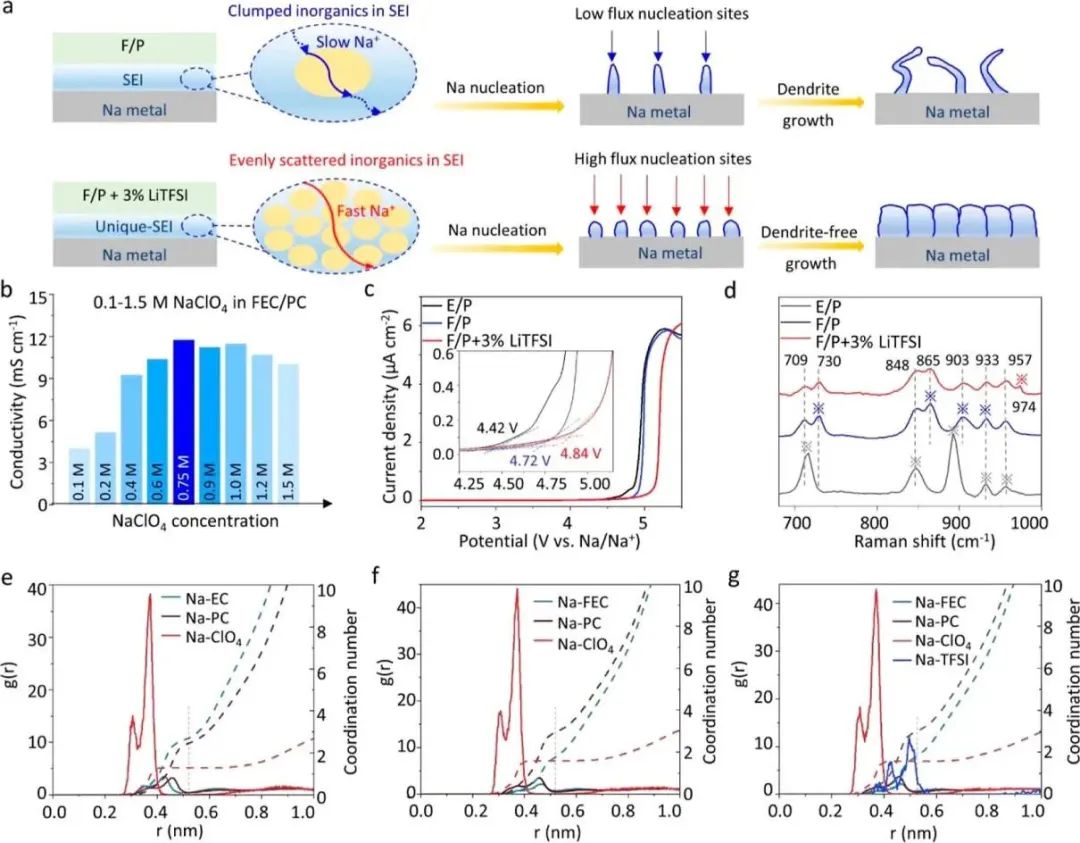
圖1( a ) F / P和F / P + 3 % Li TFSI電解液形成SEI膜的影響。( b ) FEC / PC溶劑中E / P、F / P + 3 % LiTFSI和不同濃度NaClO4的電導率。( c )對稱不銹鋼電池中E / P、F / P和F / P + 3 % LiTFSI電解液在1 m V s - 1掃描速率下的氧化穩定性。( d ) E / P、F / P和F / P + 3 % Li TFSI電解液的拉曼光譜。( e ) E / P電解液中Na - EC、Na - PC和Na - ClO4的徑向分布函數和配位數,( f ) F / P電解液中Na - FEC、Na - PC和Na - ClO4的徑向分布函數和配位數,( g ) F / P + 3 % LiTFSI電解液中Na - FEC、Na - PC、Na - ClO4和Na - TFSI的徑向分布函數和配位數。
如圖1b所示,電導率與NaClO4濃度呈下凹拋物線關系,隨著濃度的增加,在濃度低于0.75 mol·L?1之前,電導率呈上升趨勢。之后,電導率由于高濃度的NaClO4導致較高的粘度和較低的離子遷移率而下降。因此,在FEC/PC中0.75 mol·L?1的鈉鹽(記為“F/P”)被選為三種電解質的最佳濃度,其具有最高的電導率,為11.25 mS·cm?1。在將LiTFSI(3%質量比)添加到F/P后(標記為“F/P + 3% LiTFSI”),電導率略微下降至10.75 mS·cm?1。而在EC/PC中0.75 mol·L?1的NaClO4(標記為“E/P”)提供了最低的電導率,為7.16 mS·cm?1。
不同類型的電解質在圖1c中進行了測試。在基于EC的電解質中,超過4.42 V后觀察到氧化電流的增加,這可以歸因于在高電壓下EC的氧化分解。當EC被FEC替代后,氧化電流在達到4.72 V后增加。此外,在F/P + 3% LiTFSI中實現了明顯更高的氧化電壓,達到4.84 V。結果證實了LiTFSI和FEC的聯合應用能夠提高高電壓下的氧化穩定性。此外,采用拉曼光譜學來展示E/P、F/P和F/P + 3% LiTFSI電解質的溶劑化結構(圖1d)。以純FEC為參考,F/P和F/P + 3% LiTFSI電解質中的957 cm?1處的峰表示FEC對Na+的溶劑化效應。在電解質中添加LiTFSI后,865 cm?1處的峰變得較弱并且出現紅移,表明溶劑不僅與Na+具有協同作用,還與Li+具有協同作用。
陽離子和陰離子的溶劑化結構決定了電解質中離子的傳輸行為。在這里,采用分子動力學(MD)模擬來研究三種電解質中的Na+溶劑化和Li+溶劑化。如圖1e所示,通過解析E/P電解質中的徑向分布函數(RDF),可以得知Na+在其溶劑化殼中的平均配位數為2.69 EC、2.46 PC和1.28 ClO4?。在EC被FEC替代后,電子親合性較強的F基團可以減少-C=O的電子云密度,并減弱與電解質中Na+的結合能力。因此,F/P電解質中的Na+ 表現出其溶劑化殼中較低的配位數,為1.76 FEC(圖1f),導致溶劑化的Na+的快速傳輸。在引入LiTFSI后,Na+的平均配位數略微變化,而每個Na+的TFSI?配位數僅為0.022(圖1g)。不可避免地,TFSI?陰離子在電解質中是自由的,并且傾向于被吸附在鈉表面。
二 鈉金屬電池的電化學性能
除了離子導電性和擴展的物理化學性質,電化學可逆性和穩定性對于鈉金屬在電池中的應用也是關鍵因素。在這里,對三種電解質中的Na∥Al電池的恒流式長期鈉沉積和剝離行為進行了評估。
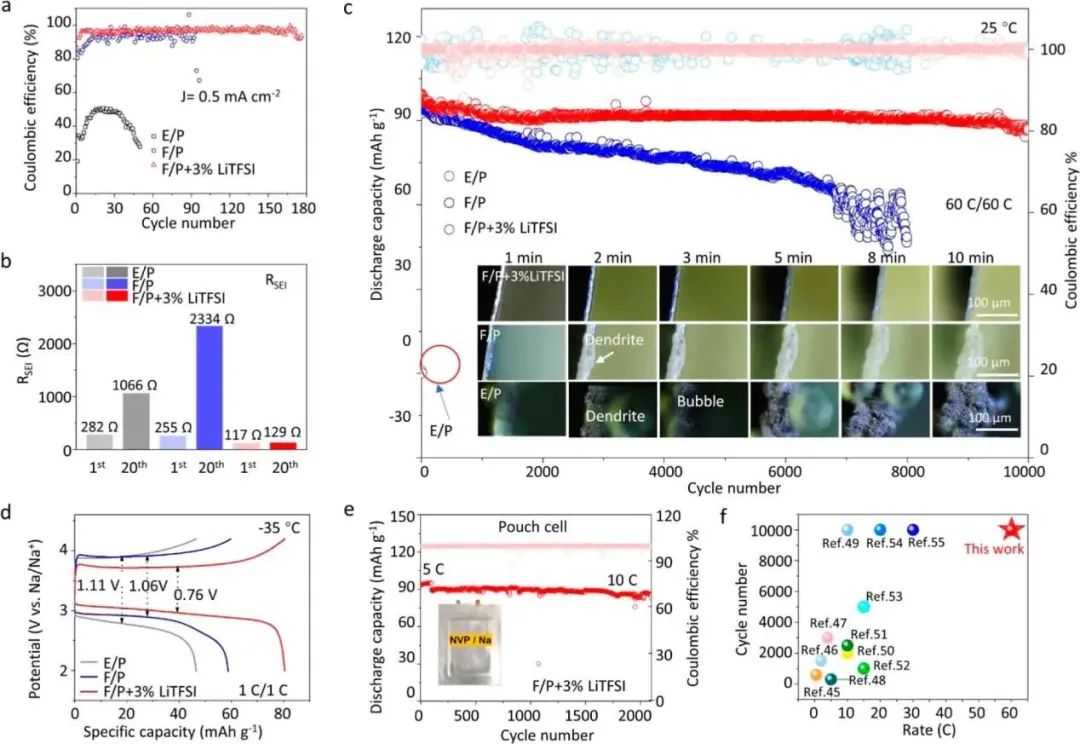
圖2 ( a ) 25 °C時,在0.5 mA·cm -2和0.5 mA·h·cm -2的電流密度下,Na∥Al半電池在三種電解液中的庫倫效率。( b ) Na∥Al紐扣電池在1和20次循環后SEI膜阻抗的擬合值。( c ) Na∥Na3V2 ( PO4 ) 3 ( NVP )電池在E / P,F / P和F / P + 3 % LiTFSI電解液中60 C / 60 C的充電/放電倍率下的循環性能;插片是原位光學顯微鏡觀察鈉枝晶在相應電解液中的生長過程。( d )在- 35 ℃下,Na / NVP電池在E / P、F / P和F / P + 3 % Li TFSI電解液中的首次充放電曲線。( e )在F / P + 3 % Li TFSI電解液中,Na / NVP軟包電池在5 C (前100個周期)和10 C倍率下的循環性能。( f )本工作中小微企業與已報道文獻的對比表現。
如圖2a所示,在E/P電解質中,50個循環的庫倫效率(CE)不足51%。而在F/P電解質中,CE提高到約80-95%,并在80個循環后變得高度波動。不穩定性歸因于SEI膜的持續溶解/再生以及鈉樹枝的不斷生長。如預期的,F/P + 3% LiTFSI電解質中的電池在近180個循環中實現了平均CE高達97.18%,表明設計的電解質中的鈉金屬具有高度可逆和穩定的性能。同時,EIS表征證實了1和20個循環后電極-電解質界面的演變。顯然,F/P + 3% LiTFSI中的阻抗在循環過程中只有相對較小的增加。通過擬合三種電解質的EIS光譜,F/P + 3% LiTFSI中的RSEI值遠低于兩種參考電解質(圖2b)。與之前的物理化學性質分析相比,SEI膜是改善CE的顯著原因。接下來,在-35°C下驗證了F/P + 3% LiTFSI電解質中SEI膜的優勢,結果表明在三種電解質中具有相對高且穩定的CE。此外,采用對稱Na/Na電池評估了三種電解質中鈉金屬電極的電化學性能,其中添加LiTFSI的電解質通過促進穩定的SEI形成和便于快速Na+傳輸動力學,支持電池實現了最長的循環時間。最后,對設計的電解質的物理化學性質進行了比較,F/P + 3% LiTFSI電解質占據了最大的面積,表明在三種電解質中具有最佳的性質。
為驗證LiTFSI在電解質中的實用性,采用Na∥NVP全電池評估了高速充放電條件下的性能。從2到100 C研究了電解質中Na∥NVP電池的速率能力。F/P + 3% LiTFSI電解質中的Na∥NVP電池可在100 C的超高速率下保持高達91.68 mA·h·g?1的容量,并且在極高的速率下仍然具有極低的極化電壓。相比之下,F/P電解質中的電池在60 C以上表現出不穩定的容量,而E/P電解質中的電池在高速率下具有急劇的容量下降。優越的速率性能得益于添加LiTFSI后形成的高度可逆和穩定的SEI。因此,在三種電解質中以60 C的充放電速率測試了Na∥NVP電池的循環穩定性(圖2c)。令人驚訝的是,使用F/P + 3% LiTFSI電解質的Na∥NVP電池在經過10,000個循環后具有出色的循環穩定性,容量保留率達到了89.15%。使用F/P電解質的參考電池在經過8000個循環后的循環保留率降至48.44%,而E/P電解質中的電池幾乎在如此高的速率下無法正常工作。比較數據表明,E/P和F/P電解質形成的SEI不夠穩固和穩定,無法支持它們在高電流率下工作,因為FEC和EC的不斷分解。此外,不穩定的SEI可能導致鈉的不均勻沉積和樹枝生長。為驗證三種電解質中的鈉沉積形態,采用Na∥Al電池的原位光學顯微鏡來展示效果。如圖2c,E/P電解質顯示出非常不均勻的鈉樹枝沉積和大量氣泡,因為EC無法形成均勻的SEI并鈍化鈉陽極。電解質中的活性鈉可能在循環性能期間耗盡,導致CE降低和電化學性能下降。在EC被FEC替代后,F/P中的鈉樹枝和氣泡減少。盡管如此,由于SEI脆弱且不均勻,電解質仍然無法防止在充電過程中鈉樹枝的生長。然而,在F/P + 3% LiTFSI電解質中,鈉樹枝和氣泡幾乎消失。這些結果表明,F/P + 3% LiTFSI電解質適用于均勻的鈉沉積。
三 SEI膜的特征
當電流通過鈉金屬時,表面上的一些陽離子會被電子消耗并還原為SEI膜的組成部分。SEI的性質,包括化學成分和結構,對Na+的傳輸產生重要影響。富含無機物的SEI已被證明具有快速離子沉積和均勻核的能力。
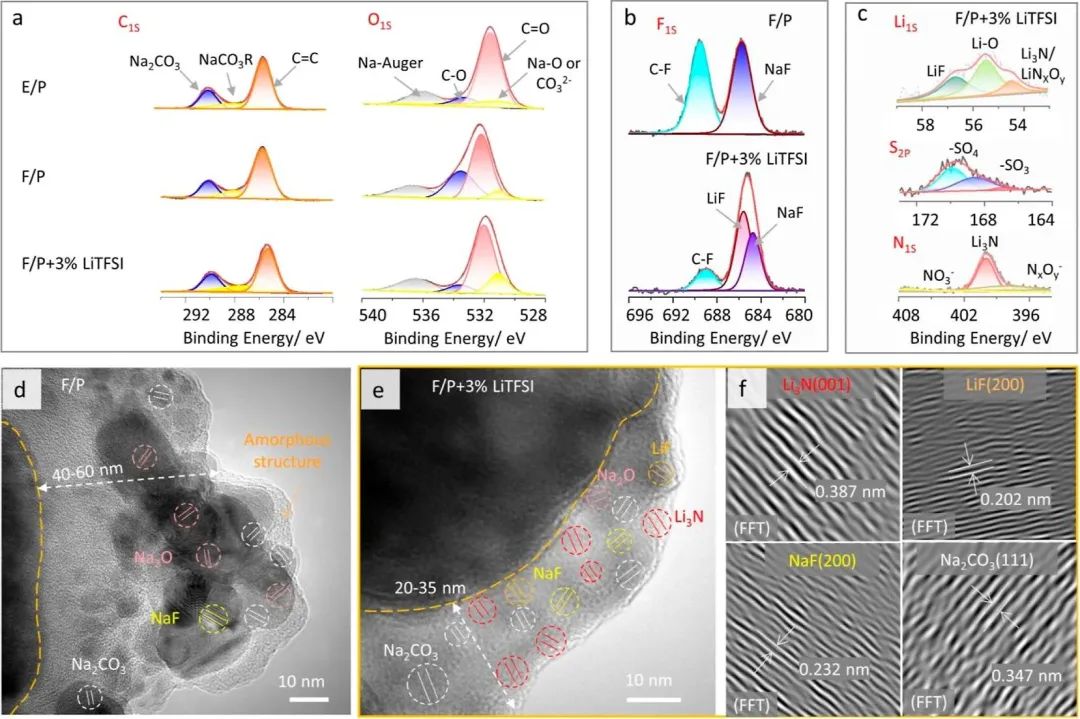
圖3 在1 mA·h·cm -2的電流下循環20次后,鋁箔基底上的SEI膜的組成和相應的XPS圖譜由( a )式表示。E / P、F / P和F / P + 3 % Litfsi電解液中的C1S和O1S;( b ) F / P和F / P + 3 % LiTFSI電解液中的F1S;( c ) Li1s、S2p和N1s在F / P + 3 % Li TFSI電解液中,在相應電解液中反復鍍/脫Na后Ar +濺射。( d ) F / P電解液和( e ) F / P + 3 % Li TFSI電解液中循環3次后SEI膜的TEM成像。( f )通過傅里葉變換( FT )從圖3e中得到SEI的晶格條紋圖像。
因此,通過XPS對三種電解質形成的SEI膜的組成進行了表征(圖3a)。在三種電解質中都檢測到Na2CO3、NaCO2R、Na2O等無機組分。事實上,這些組分不能阻止鈉表面上的副反應,并導致E/P電解質的電化學性能差。在用FEC替換EC后,F/P和F/P + 3% LiTFSI電解質中的F1s光譜顯示了NaF(684.7 eV)和C?F(690 eV)峰(圖3b)。盡管NaF可以保護鈉金屬并在循環過程中防止樹枝生長,但當含量過高時,它被證明不是一個理想的組分,可能會阻礙Na+的傳輸。而引入LiTFSI到電解質中后,檢測到了強烈的685.5 eV處的峰,應該來源于LiF。由于其較高的離子導電性,LiF對于降低界面的電阻更為有效,超過了NaF,它可以阻止電子的穿隧并防止電解質的分解,導致可逆鈉沉積和剝離過程中更薄的SEI。在循環過程中,LiF還可以抑制垂直樹枝的生長,因為其約64.9 GPa的高楊氏模量。此外,C?F峰變得較弱,說明LiTFSI可以防止FEC的分解,這也反映在原位電子顯微鏡中,對應較少的氣體和樹枝生長。此外,SEI膜中的高導電組分是由TFSI?形成的(圖3c),400 eV處的峰可以歸因于N3?或NxOy含鹽,169 eV是SO42?鹽,56.5 eV是LiF,168 eV是SO32?鹽。N含鹽有資格改善Na+的傳輸和SMBs的高速率性能。與參考電解質相比,F/P + 3% LiTFSI電解質中的元素不同且多分散;某些新元素,如Li/N/S/F,在SEI中的份額分別為4.48、1.56、0.69和6.15%(原子%)。總體而言,F/P + 3% LiTFSI中的無機成分高于其他兩種電解質,從而導致較高的電導率。
進一步使用透射電子顯微鏡(TEM)對這些無機和有機成分的詳細結構和分布進行了表征。如圖3d所示,F/P電解質中SEI的組分主要由大尺寸和笨重的無機物組成,例如Na2O、Na2CO3、NaF等,以及外層豐富的無定形有機物。盡管無機物由于其親鈉性能可以引發快速的Na+擴散,但有機物外層由于其導電性差而限制了這一過程,從而限制了高流動性Na+的沉積并導致樹枝的生長。相反,在F/P + 3% LiTFSI中SEI的無機物(圖3e)呈點狀分布,厚度僅為20-35 nm(與F/P電解質中40-60 nm的厚度相比),可以形成快速的Na+域和高通量的成核點。短程無機位點可以降低Na+的傳輸能量,并促進快速Na+沉積。更有趣的是,傅里葉變換的SEI中的晶格間距圖像(圖3f)確定了Li3N(001)、LiF(200)、NaF(200)和Na2CO3(111),圖S22也證明了這些無機物的相關晶格間距。在這些無機物中,高導電性的Li3N均勻地點綴在整個SEI的外層和內層,這將降低界面和SEI內部活性離子的傳輸能量壁壘,有利于Na+的均勻沉積和快速傳輸。
四 SEI膜的形成機制
為了研究電解質中每個組分的單獨效應,通過DFT計算了溶劑和鹽的最高占據分子軌道(HOMO)和最低未占據分子軌道(LUMO)能級。
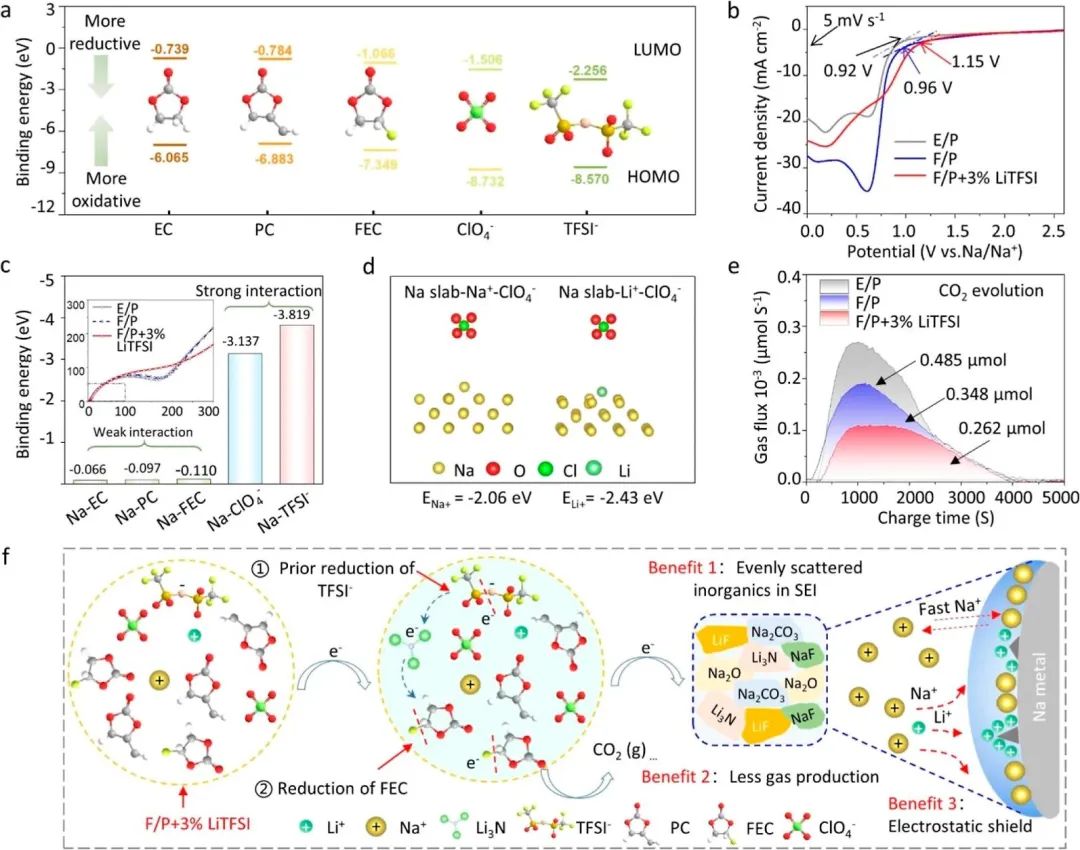
圖4 ( a )通過密度泛函理論( DFT )計算了EC,PC,FEC,ClO4 -和TFSI -的LUMO能級。( b ) Na / Al電池在E / P、F / P和F / P + 3 % LiTFSI電解液中的線性掃描伏安(LSV)曲線。( c ) Na - EC、Na - PC、Na - FEC、NaClO4-和Na - TFSI-的結合能。(插圖為組裝后Na / Al扣式電池在3種電解液中的阻抗譜。) ( d )Na-Na +和Na - Li +的結合能。( e ) Na / Al電池在3種電解液中的析CO2量。( f ) F / P + 3 % Li TFSI電解液分解機理示意圖及帶來的好處。
如圖4a所示,TFSI?的LUMO能級低于ClO4?,表明其更傾向于在陽極表面還原并參與SEI的形成。與EC和PC相比,FEC的LUMO能級較低,也使其參與了SEI膜的形成。至于圖4b中的線性伏安掃描,三種電解質中的Na∥Al電池在0?2.8 V的電壓范圍內進行了測試,有助于我們了解TFSI?在鈉金屬IHP中的特定吸附和還原過程。這些曲線顯示,具有LiTFSI(F/P + 3% LiTFSI)的電解質還原電位從1.15 V開始,早于F/P(0.96 V)和E/P(0.92 V)電解質中的電位,表明了添加劑LiTFSI的優先還原和自我犧牲。為了進一步驗證LiTFSI優先還原的原因,通過DFT計算了電解質中鈉金屬對陰離子的吸附能量(圖4c)。能量值證實TFSI?的吸附能量高于ClO4?(-3.819 eV對-3.137 eV),表明TFSI?更傾向于吸附在鈉金屬表面上,從而積極參與SEI膜的形成。與此同時,電阻測試(圖4c中的插圖)在三種電解質中的新組裝的Na∥Al電池上進行,結果顯示E/P和F/P具有類似的阻抗曲線。由于來源于IHP的阻抗,這些阻抗與鈉金屬表面吸附的離子種類有關,這兩種電解質中的吸附在鈉金屬上的離子相似。然而,F/P + 3% LiTFSI中的電池曲線與其他兩者不同,表明引入LiTFSI導致了鈉金屬吸附離子的變化(對應于TFSI?)。圖4d顯示,Na?Li+的結合能高于Na?Na+,這意味著電解質中的Li+更傾向于吸附在鈉金屬表面上,并參與SEI膜的形成。
與F/P電解質中的吸附離子相比,F/P + 3% LiTFSI中的TFSI?離子和Li+更傾向于吸附在鈉金屬的IHP上并參與SEI的形成。優先吸附的優勢也通過活化能(Ea)計算得到了確認。通常,Na+必須克服電解質和電極之間的相界面的能量壁壘;通過不同溫度下的電化學阻抗譜(EIS)可以確定此過程的RSEI的Ea。在F/P + 3% LiTFSI中計算得到的Ea(SEI)的絕對值為9.93 kJ·mol?1,低于E/P和F/P電解質中的值,表明Na+更有可能通過由LiTFSI誘導的IHP形成的SEI膜。
來自添加LiTFSI的電解質的SEI的優勢也在循環伏安(CV)測試中表現出來的Na+鍍/剝離動力學行為中提供。F/P + 3% LiTFSI電解質中的鈉鍍覆過電位為28 mV,遠低于F/P(59 mV)和E/P(67 mV)電解質的值。在CV的正掃描中,F/P + 3% LiTFSI電解質中的Na/Al電池顯示出比其他兩者更高的峰面積,表明了鈉鍍/剝離的最高容量和最佳逆轉性。結果表明F/P + 3% LiTFSI電解質對Na+傳輸具有最佳的動力學性能。
析氣問題是SMB中許多碳酸鹽電解質所面臨的障礙之一。根據上述討論,自我犧牲型的LiTFSI導致的獨特SEI對加速Na+傳輸和調控Na+沉積產生了良好的促進作用。我們認為這種膜也可能解決氣體問題。通過原位氣相色譜-質譜儀(原位GC?MS)檢測了在放電過程中三種電解質中產生的氣體組分。結果表明,這三種電解質中產生的主要氣體是H2和CO2。H2主要來自鹽中水分與鈉金屬反應,而CO2主要來自EC或FEC的分解。從圖4e中可以看出,E/P具有最高的氣體產量,為0.485 μmol,主要歸因于EC的分解。F/P由于FEC的分解,氣體產量較低,為0.348 μmol。然而,F/P + 3% LiTFSI的氣體產量僅為0.262 μmol,表明LiTFSI的添加可以抑制FEC的分解并減少CO2的生成。
圖4f說明了F/P + 3% LiTFSI電解質中SEI的形成機制;LiTFSI的優先還原產生了Li3N中的孤電子對,它充當活性中心,并催化了FEC的還原。因此,添加LiTFSI可以為電池帶來三個好處,如均勻分散的獨特SEI中的無機物、較低的CO2氣體生成以及來自Li+的靜電屏蔽(不參與SEI的形成)。在其中,NaF/LiF等鹽由于其較高的楊氏模量在阻止樹枝生長方面是積極的。毫無疑問,富含N的鋰鹽(如Li3N和LiNxOy)還提供了高導電性,并有利于Na+的傳輸,促進了鈉的均勻沉積。有利的SEI不僅可以減輕電解質與鈉金屬之間的有害反應,還可以促進高速率下的高效Na沉積。
為了進一步闡明在通過添加LiTFSI形成的SEI中增強的Na+鍍/剝離動力學,推斷了反應機制。由于鈉金屬的吸附,Li+和TFSI?首先引入IHP并參與SEI的形成,產生富含Li3N的無機SEI膜。然后,Li3N充當“種子”催化了FEC的還原。由于濃度和還原動力學抑制了LiTFSI的分解,反應較溫和,分解產物多點生成,并分散在Li3N周圍。由于FEC的還原可以產生大量的無定形有機物,因此像Li3N、LiF和NaF等無機物也會交叉生成并分散存在。最重要的是,還原競爭反應在SEI中產生較少的有機物,尤其是在外層。此外,高導電性的Li3N保證了高通量的成核位點,而NaF/LiF在高速循環性能中促使相界面保持穩定。
五 CEI的特征及成分分析
為了研究電解質對NVP陰極材料的相互作用和影響,采用Cryo-TEM在循環過程中測量了CEI的特性。
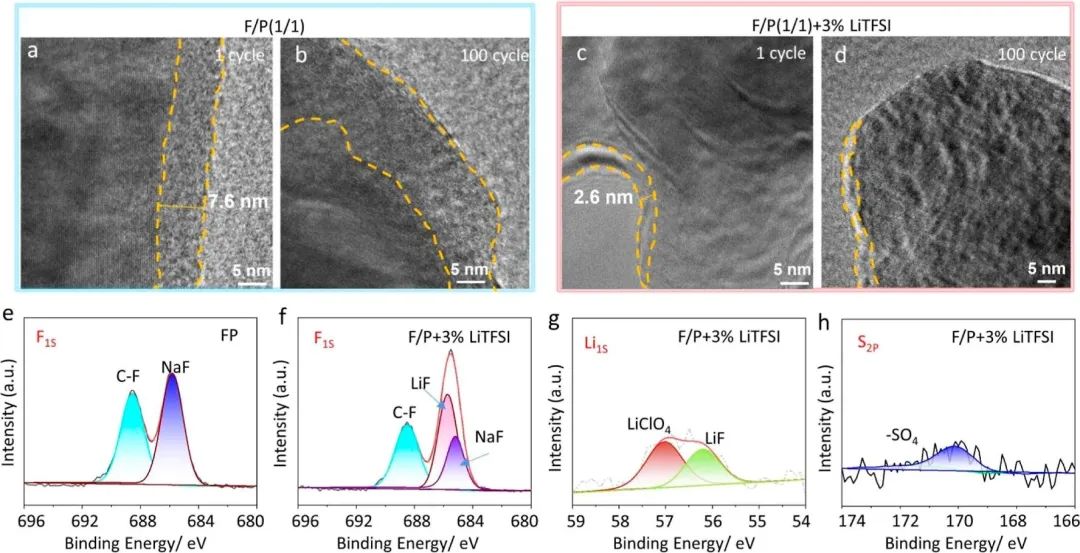
圖5 Na3V2 (PO4) 3正極在F / P電解液中經過( a ) 1次循環和( b ) 100次循環,在F / P + 3 % Li TFSI電解液中經過( c ) 1次循環和( d ) 100次循環后的低溫TEM表征。Na∥Na3V2 (PO4) 3電池在30 C下循環100次后Na3V2 (PO4) 3基底上CEI膜的組成以及重復Na +鍍/脫后Ar +濺射F / P中( e ) F1s和F / P + 3 % Li TFSI中( f ) F1s、( g ) Li1s和( h ) S2P對應的XPS圖譜。
在第一次循環后,F/P中NVP陰極上的CEI約為7.6 nm厚(圖5a),變得均勻,在100次循環后厚度增加到約20 nm(圖5b)。相比之下,F/P + 3% LiTFSI電解質中的CEI在第一次循環后僅約為2.6 nm厚(圖5c),即使在100次循環后仍然均勻而光滑,厚度約為5 nm(圖5d)。從這些結果可以清楚地看出,LiTFSI可以在循環過程中生成穩定而堅固的CEI。此外,經過一個循環后的NVP顆粒的SEM圖像證實,LiTFSI可以抑制顆粒在重復插入/提取鈉反應過程中的損傷。
XPS被用來研究F/P和F/P + 3% LiTFSI電解質中循環后NVP的CEI膜。這兩種電解質中CEI的共生產物主要由C?F、C?C、C?H、C?O以及一些由碳酸鹽溶劑分解產生的Na2CO3、ROCO2Na和Na2O組成。由于這兩種電解質中存在FEC,因此在陰極上形成了C?F峰和NaF(圖5e,f)。當引入LiTFSI到電解質中時,發現C?F峰的強度較弱,表明抑制了FEC的分解。圖5g中的Li1S峰顯示CEI膜中含有一定量的LiF,其來源于LiTFSI和FEC的分解。而57 eV歸因于LiClO4,170 eV處的S2p峰(圖5h)起源于TFSI?中的SO42?。這些多種無機層限制了電解質的進一步分解,提高了CEI膜的穩定性。此外,與NaF相比,后者的Na+導電性較差,而LiF則具有較高的導電性,有利于減小界面阻抗的降低。因此,引入LiTFSI到電解質中有利于降低CEI膜的阻抗并增強其動態性能。需要注意的是,CEI膜中沒有含氮組分。此外,CV測試說明了LiTFSI的添加在循環過程中引起了動態變化;然而,在隨后的循環過程中它們迅速變得穩定。因此,混合型CEI可以維持堅固而緊湊的保護層,有效抑制了大多數陰極-電解質相界面的副反應,從而使電池具備高速率和長周期性能。
總結與展望
在這項工作中,精心設計了一個SEI,其中分散的無機物有助于提高SMBs的超高速率和長壽命。電化學測試揭示了LiTFSI在促進Na∥NVP電池在60 C下循環并在經過10,000個循環后保持89.15%容量保留方面發揮了至關重要的作用。這種SMBs的電化學結果優于先前報道的性能。結合XPS、拉曼、低溫透射電子顯微鏡(Cryo-TEM)和理論計算,我們發現Li+和TFSI?可以優先吸附在鈉金屬的IHP層,并首先還原為Li3N,這證明了活性中心并催化了FEC的還原。因此,LiTFSI和FEC之間的還原競爭效應促進了SEI的形成,其中均勻分散的高導電性無機物,為Na+提供了快速的傳輸通道和高通量成核位點,實現了高速率下的快速鈉沉積。獨特的SEI膜還抑制了FEC的分解并減少了氣泡問題。與此同時,LiTFSI還有助于形成堅固而緊湊的CEI膜。這種點綴有無機物的保護性界面層策略為開發超高速率和長壽命的SMBs以及其他金屬電池打開了新的途徑。
審核編輯:劉清











 工商網監
工商網監
評論