1
研究背景
室溫鈉硫電池因其理論能量密度高、成本低、資源豐富、環境友好等優點而受到人們的廣泛關注。但它仍然面臨以下挑戰:(1)硫正極及其放電產物的低電導率,反應動力學緩慢,多硫化鈉(NaPS)的穿梭效應。(2)Na枝晶的形成。由活性中心組成的極性納米結構材料能有效吸附極性NaPS,并可作為電催化劑,加速NaPS轉化為Na2S。
但是,低密度的催化活性位點和過量的金屬負極會抵消Na-S電池的高能量密度優勢。另外,在Na金屬負極中,輕質多孔碳材料由于其優異的導電性和化學穩定性,有望抑制Na枝晶形成。然而,大多數碳骨架通常是疏鈉的。因此,針對高能量密度Na-S電池體系,研究一種能同時調節陽離子和陰離子遷移行為的協同策略具有重要意義。
2
成果簡介
近日,清華大學李亞棟院士和王定勝副教授聯合中科院福建物質結構研究所溫珍海教授在JACS上發表了題為“Single-Atom Yttrium Engineering Janus Electrode for Rechargeable Na–S Batteries”的論文。該論文采用金屬-有機框架(MOF)制備了一個單原子雜化物,其中Y單原子被引入氮摻雜的四邊形碳宿主(Y SAs/NC)中,該雜化物具有良好的親鈉和親硫Janus性質,因此當用作鈉-硫全電池的鈉負極和硫正極的宿主時具有良好的電化學性能。
該Na-S全電池能夠提供822 mAh g-1的高容量,并顯示出優異的循環穩定性(在5 A g-1的下,循環超過1000圈后的容量保留率為97.5%)。3D打印電池和Na-S軟包電池進一步驗證了這種Na-S電池的實際應用潛力。
3
研究亮點
(1)理論計算表明,YN4/C可以降低Na2S的分解能壘,表現出較強的多硫化物(Na2S6)吸附能,并能夠提供有效的親鈉位點,促進鈉均勻成核。 (2)因此,將Y單原子引入到N摻雜碳多面體(Y SAs/NC)中,構建了Y SAs/NC-S||Y SAs/NC-Na全電池,該電池具有高容量、優異的倍率性能和循環穩定性。 (3)機理研究表明,原子Y可以電催化S8還原為Na2S,加速反應動力學,有效緩解“穿梭效應”,并在高電流密度下表現出穩定的循環性能。
4
圖文導讀
首先,通過DFT計算預測了YN4/C的電催化SRR動力學和Na沉積行為。降低Na2S的分解能壘可以極大地促進活性材料的利用,減少非活性Na2S的形成,從而達到較長的循環壽命。因此,系統地計算了Na2S在不同基底和N摻雜石墨烯(NC)上的分解能壘。圖1a顯示,與MnN4/C(1.54 eV)、ZnN4/C(1.84 eV)、NiN4/C(1.93 eV)、NC(2.00 eV)、CuN4/C(2.02 eV)、FeN4/C(1.87 eV)和CoN4/C(1.85 eV)相比,YN4/C在充電過程中對Na2S具有最小的分解勢壘(1.52 eV),表明其能夠促進Na2S的催化分解。
此外,Na2S6與YN4/C之間的結合能為?2.30 eV(圖1b),明顯強于Na2S6在其他基底上的結合能,這有利于硫還原反應并抑制多硫化物穿梭。圖1c顯示,YN4/C的電子離域比NC的要強烈得多。在YN4/C中,適度的電子離域可以降低電荷轉移電阻,有利于均勻的金屬沉積。
圖1e的模擬鈉沉積過程顯示,在NC上,Na簇早在300ps就已經形成,而在YN4/C上即使在500ps也不會產生(圖1d)。圖1f顯示了高度配位的Na原子數隨時間的變化。圖1g,h的電荷密度差分析顯示,與NC相比,由YN4/C區組成的區域Na+與YN4/C之間的相互作用更強,說明Na+在這些活性位點上被有效吸收,協同促進了Na+的存儲性能。圖1i顯示,Na從石墨烯晶格向Y中心的遷移以放熱為主,說明這是一個自發過程。 ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ?
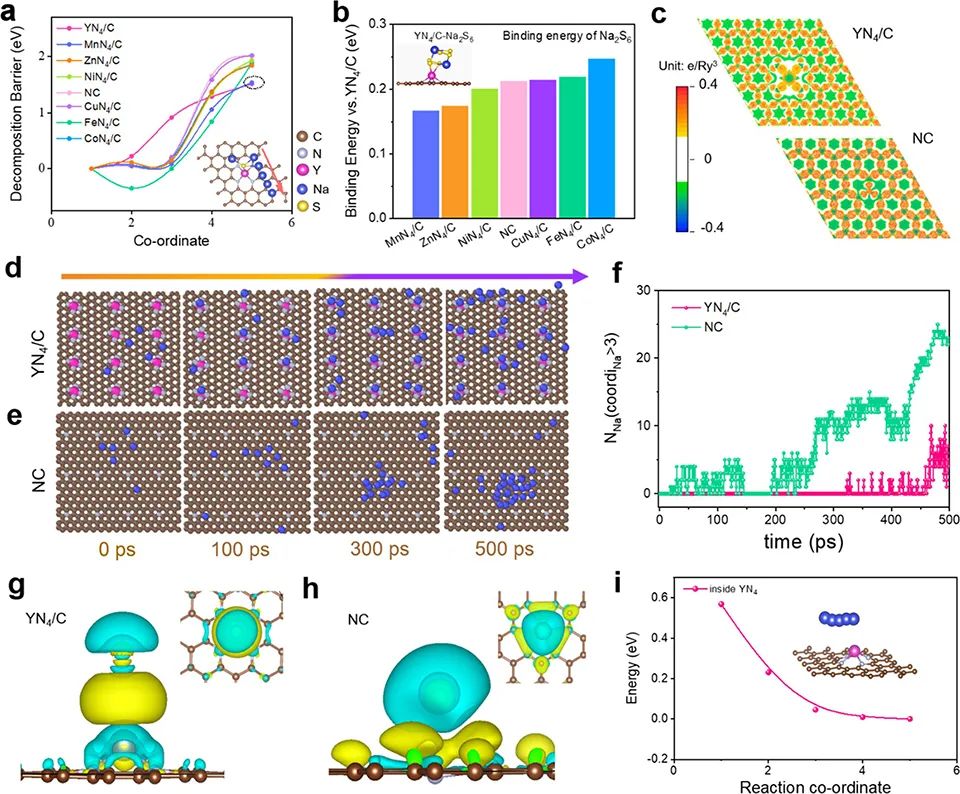
圖 1、(a)Na2S在YN4/C、MnN4/C、ZnN4/C、NiN4/C、NC、CuN4/C、FeN4/C、CoN4/C等不同底物上的分解勢壘。(b)相對于YN4/C,Na2S6在MnN4/C、ZnN4/C、NiN4/C、NC、CuN4/C、FeN4/C、CoN4/C上的吸附能。(c)YN4/C和NC的電子局域函數圖。YN4/C(d)和NC(e)電極在特定模擬時間下的分子動力學模擬快照。(f)有3個以上Na配位的Na原子數隨時間的變化。Na在YN4/C(g)和NC(h)電極上的吸附電荷密度差的俯視圖和側視圖。(i)Na從石墨烯晶格遷移到YN4基團的擴散能壘。
從Y SAs/NC的TEM(圖2a)可以看出,合成后的Y SAs/NC結構很好地保留了Y(acac)3/ZIF-8前驅體的初始菱形十二面體形態和尺寸分布。經像差校正的高角度環形暗場掃描透射電鏡(AC-HAADF-STEM)圖像(圖2b)顯示,存在高密度的單個亮點 (圖2c),證明了Y在骨架上的原子級分散。能量色散X射線能譜圖(EDS)顯示,Y、C和N在整個十二面體中均勻分布(圖2d)。
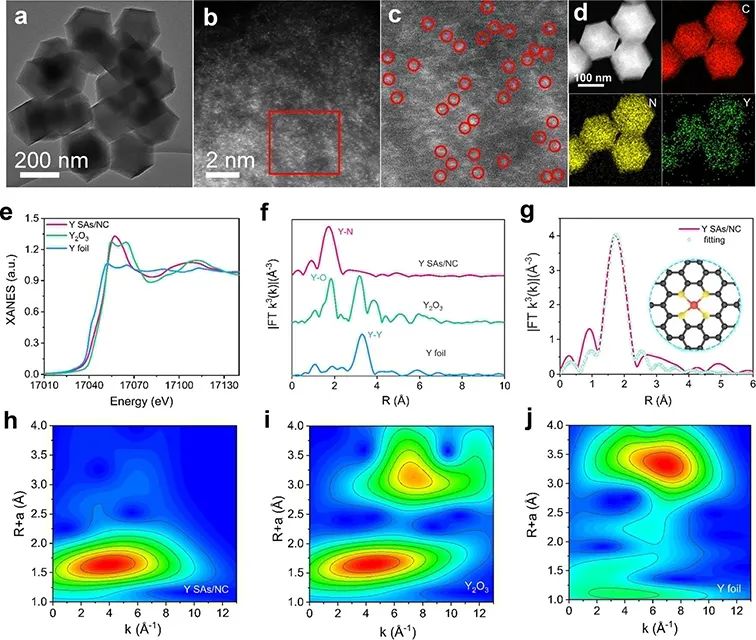
圖 2、Y SAs/NC的TEM圖像(a)和AC-HAADF-STEM圖像(b)。(c)放大AC-HAADF-STEM圖像。(d)Y SAs/NC的EDS元素映射。(e)不同樣品的Y K邊X射線吸收近邊結構(XANES)光譜和(f)傅里葉變換(FT)k3加權擴展X射線吸收精細結構(EXAFS)光譜。(g)Y SAs/NC的EXAFS擬合圖。(h-j)Y SAs/NC、Y2O3和Y箔的小波變換-EXAFS(WT-EXAFS)圖。
進行了軟X射線吸收近邊結構(XANES)測量,以確定Y SAs/NC中N和C的電子結構。Y K邊XANES譜的閾值能量位于Y箔和Y2O3的閾值能量之間(圖2e),說明Y在Y SAs/NC中的平均氧化態約為2.31。圖2f中Y SAs/NC的FT-EXAFS分析顯示,在1.71 ?附近有一個主峰,這是由于第一殼層Y-N路徑的散射。
此外,與Y箔相比,Y SAs/NC中未檢測到主峰在3.28 ?左右的Y-Y鍵,說明Y原子分散在碳基體上,具有Y-N配位。 Y SAs/NC的配位構型為一個完美平面YN4配位,如圖2g所示。
此外,在k和R空間中進行了高分辨率的小波變換EXAFS(WT-EXAFS),以識別Y SAs/NC中Y物種的原子分散狀態。如圖2h–j所示,Y SAs/NC的WT計數曲線在3.9°-1左右出現最大信號,位于Y箔(Y–Y,6.8°-1)和Y2O3(Y–O,4.3°-1)之間,表明Y–N鍵在Y SAs/NC中占主導地位。
圖3a 顯示,在1.0 mA cm-2@1.0 mAh cm-2下,Y SAs/NC能夠循環400次,具有穩定的庫侖效率(CE),超過99%。而NC和裸Cu的CE波動較大,這主要是由于Na金屬負極表面沉積了Na枝晶或死Na。圖3b顯示,Y SAs/NC-Na在三個電極中極化最小。在1.0 mA cm-2@1.0 mAh cm-2下,Y SAs/NC的成核過電位低至22.4 mV,低于NC(32.9 mV)和Cu(48.4 mV)電極的成核過電位,表明Y SAs/NC具有良好的親鈉性,能夠實現均勻的Na沉積動力學。
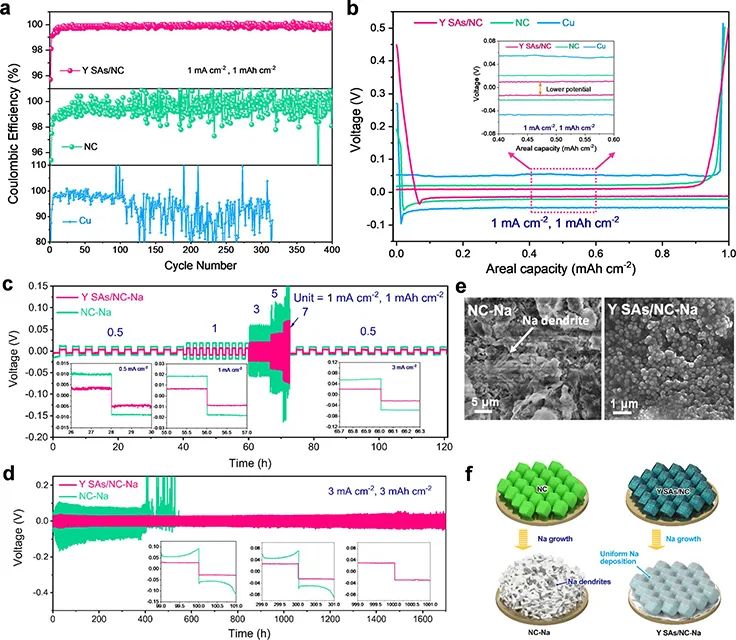
圖 3、(a)1.0 mA cm-2@1.0 mAh cm-2下,不同電極的庫侖效率和(b)電壓分布。(c)Y SAs/NC-Na|Y SAs/NC-Na和NC-Na|NC-Na對稱電池倍率性能。(d)Y SAs/NC-Na和NC-Na對稱電池長循環穩定性。(e)循環后的NC-Na和Y SAs/NC-Na負極SEM圖像。(f)Na在NC和Y SAs/NC電極上的電鍍行為示意圖。
圖3c顯示,Y SAs/NC-Na電極表現出穩定的倍率性能和較低的極化,而在5 mA cm-2時,NC-Na對稱電池產生了巨大的電壓波動。圖3d顯示,在3 mA cm-2@3 mAh cm-2下,Y SAs/NC-Na電極能夠穩定循環1500 h,而NC-Na電極只能循環小于550 h(圖3d)。
圖3e顯示,在3 mA cm-2@3 mAh cm-2循環后,NC-Na電極形成了明顯的“空腔”和Na枝晶。而Y SAs/NC沒有觀察到Na枝晶。圖3f顯示,金屬Na以均勻顆粒的形式沉積在Y SAs/NC-Na復合材料上,這得益于YN4位點的調節作用。
圖4a的循環伏安(CV)曲線顯示,Y SAs/NC-S正極有兩個清晰的陰極峰,對應于固態硫還原為可溶的長鏈NaPS(Na2Sx, 4<x<8,峰a在1.56V處)和隨后向不溶性Na2S2/Na2S的轉化(峰b在1.11 V處)。陽極峰(峰c在1.86 V處)對應Na2S到NaPS的轉化。
Y SAs/NC-S的陽極和第二個陰極峰之間的電壓差為0.26 V,明顯低于NC-S的0.34 V極化,表明YN4基團抑制了電化學極化,改善了轉化動力學。
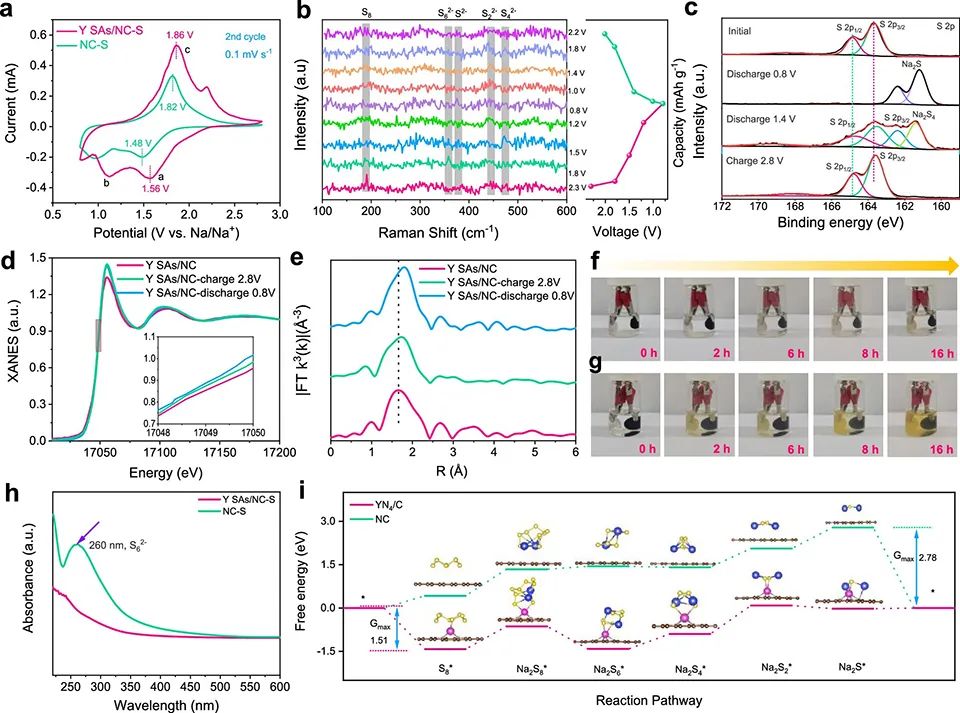
圖 4、(a) 在0.1 mV-1 s-1下,Y SAs/NC-S和NC-S正極的CV曲線。(b)由Y SAs/NC-S組成的室溫Na-S電池原位拉曼光譜。(c)Y SAs/NC-S正極在不同充放電狀態下的非原位XPS光譜。(d)在0.1 A g-1下,Y SAs/NC-S在原始狀態和不同充放電狀態下的Y K邊XANES光譜。(e)不同狀態下對應的Y K邊傅里葉變換(FT)k3加權EXAFS光譜(R空間)。從初始狀態到不同放電狀態,Y SAs/NC-S(f)和NC-S的顏色變化。(h)16 h放電循環后Y SAs/NC-S和NC-S的紫外-可見(UV-vis)光譜。(i)計算Y SAs/NC和NC上S物種逐步還原的吉布斯自由能。
圖4b的原位拉曼光譜顯示,當電池放電至1.8 V時,190.7 cm-1處的S拉伸振動帶消失,并出現另一個峰(≈358 cm-1),該峰值可對應Na2S6。在隨后的放電過程中,Na2S4在約444 cm-1處進一步轉化為Na2S2。放電結束時,Na2S在≈375 cm-1處形成。
隨后,Na2S氧化為Na2Sx(1.8 V),然后Na2Sx依次轉化為長鏈多硫化鈉(2.2 V),最后在Y SAs/NC催化下可逆轉化為S。 圖4c的XPS顯示,在放電至1.4 V時,檢測到位于161.4和162.4 eV的雙峰,對應Na2S4。在163.6和164.8 eV處產生了新的峰,這可以歸因于Na2S。這表明Y SAs/NC-S具有優越的反應動力學。
在Y K邊XANES光譜中(圖4d),由于氧化Y態的還原,Y SAs/NC-S中Y元素的K吸收邊在放電狀態下向能量較初始Y SAs/NC低的方向移動。在隨后的充電過程中,吸收邊逐漸移回到更高的能量,說明絡合物轉移回了原始狀態(圖4e)。這證實Y SAs/NC-S表面上的鍵合NaPS失去電子并轉化為硫,因此,Y位的還原狀態也被可逆地氧化。
圖4f顯示,在Y SAs/NC-S電池中,電解液在放電16小時后仍然是無色的,而在NC-S電池中,電解液在放電2小時后變為微淡黃色(圖4g)。這歸因于YN4位點對可溶性多硫化鈉的吸附。UV-vis光譜(圖4h)顯示,Y SAs/NC中Na2S6的強度下降(峰在260 nm處),證實了NaPS與Y單原子之間有很強的化學親和力。
圖4i計算的基于S8和Na可逆生成Na2S的整個反應能量路徑顯示,在放電過程中,首先是S8吸附,然后S8與兩個Na+雙還原生成Na2S8,隨后Na2S8進一步還原,形成Na2S6、Na2S4、Na2S2三個NaPS中間產物,最終生成Na2S。對于NC,由于吸附相當弱,沒有發現占主導地位的吸附物質,而Na2S*是決速組分。
對于YN4/C, Na2S6*和Na2S2*分別為主要吸附組分和決速組分。NC和YN4/C的Gmax分別為2.79和1.51 eV,證實Y SAs/NC上的硫還原過程比NC上的更容易。 將Y SAs/NC Na負極和Y SAs/NC-S正極配對組裝Na–S全電池(圖5a)。圖5b顯示,Y SAs/NC-S||Y SAs/NC-Na表現出優越的倍率性能。此外,在5 A g-1下,經過1000次循環后,其容量達到510 mAh g-1,高于Y SAs/NC-S||Na的容量(圖5c)。
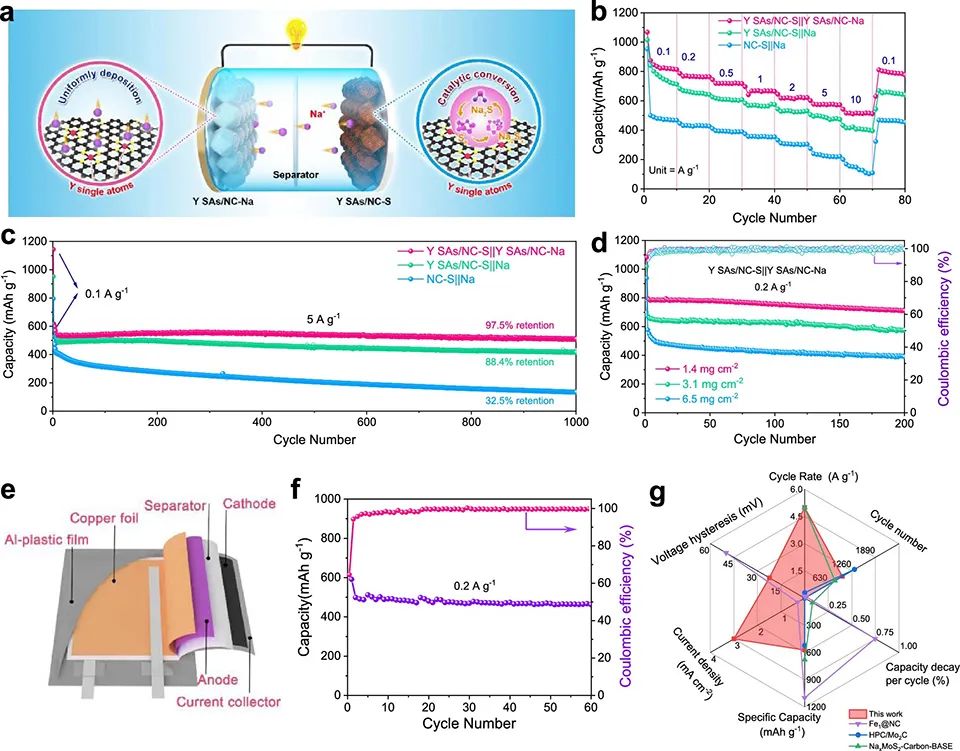
圖 5、(a)Y SAs/NC-S||Y SAs/NC-Na全電池示意圖。(b)Y SAs/NC-S||Y SAs/NC-Na, Y SAs/NC-S||Na和NC-S||Na全電池倍率性能。(c)在5 A g-1下,不同全電池的長循環穩定性。(d)Y SAs/NC-S||Y SAs/NC-Na全電池與3D打印Y SAs/NC-S||Y SAs/NC-Na全電池循環性能的比較。(e)具有Y SAs/NC-S正極的柔性Na-S軟包電池示意圖。(f)Na-S軟包電池在0.2 A g-1下的循環性能。(g)Y SAs/NC-S||Y SAs/NC Na電池與一些最先進的Na-S體系的比較。
此外,Y SAs/NC-S||Y SAs/NC Na全電池在0.2 A g–1下可提供787 mAh g–1的容量(圖5d)。載量為3.1和6.5 mg cm–2的3D打印全電池在0.2 A g–1下第五次循環容量分別為656和522 mAh g–1。即使在0.2 A g–1下,6.5 mg cm–2硫載量的全電池進行200次循環后,仍能保持近100%的庫倫效率,且每個循環的衰減率低,為0.33%(圖5d)。
此外,還組裝了一個Y SAs/NC-S軟包電池(圖5e)。將軟包電池載量增加至~80 mg(對應~2.3 mg cm–2)。圖5f顯示,在0.2 A g–1下,組裝后的電池容量高達500 mAh g–1。圖5g顯示,Y SAs/NC-S||Y SAs/NC Na性能優于一些最先進的Na–S體系。
5
總結與展望
本工作設計了基于Y SAs/NC-S正極和Y SAs/NC-Na負極的新型Na-S全電池。理論預測表明,YN4/C具有優化的電荷結構和可調的電子局域化特征,可以促進均勻的Na沉積,防止枝晶生長,加速NaPS轉換,降低Na2S分解能壘。Y SAs/NC作為同時調節S正極和Na負極的Janus功能宿主,對促進S氧化還原反應動力學和抑制多硫化物穿梭過程具有協同作用,抑制了Na枝晶生長。
構建的Y SAs/NC-S||Y SAs/NC-Na全電池在0.1 A g-1下具有822 mAh g-1的容量。此外,采用Y SAs/NC-S正極的軟包電池在0.2 A g-1下具有500 mAh g-1的高面積容量,證明了其在柔性Na-S電池中的實際應用前景。本工作為制備高性能Na-S全電池提供一種實用的策略。
審核編輯:劉清